Дипломная работа: Поиск новых фторидофосфатов лития и переходных металлов
Сложный оксид LiCoO2 обладает слоистой структурой, в которой ионы лития и кобальта упорядочены в чередующихся плоскостях. Наличие плоскостей, занятых исключительно ионами лития, обеспечивает возможность почти полного извлечения щелочного металла и тем самым применимость данного соединения в качестве катодного материала в химических источниках тока. Но продукт полного извлечения – слоистый CoO2 – очень неустойчив, и на практике циклирование ведут в диапазоне x от 0 до приблизительно 0,5[2].
При десяти циклах заряд (4,2 В) - разряд (3,5 В) начальная удельная разрядная емкость 145 A*час/кг. Потери разрядной удельной емкости 0,1% на 1 цикл [5].
К недостаткам кобальтита лития относят то, что при многократном циклировании часть ионов кобальта перемещается в литевые слои, слоистая структура перестраивается в каркасную типа шпинели, и движение ионов лития затрудняется, а также высокую стоимость и токсичность [2].
Поэтому ведутся интенсивные поиски и исследования альтернативных материалов. В частности, большое число работ посвящено легированию LiCoO2 , структурно родственным ему соединениям LiMnO2 , LiNiO2 , фазам типа шпинели на основе LiMn2 O4 и др. В частности, хорошо зарекомендовали себя фазы типа оливина LiMPO4 (где M = Mn, Fe, Co, Ni), описываемые ниже.
1.2. Смешанные фосфаты лития и переходных металлов
Двойные фосфаты, имеющие общую формулу LiMPO4 (где M = Mn, Fe, Co, Ni), изоструктурны оливину - силикату магния и железа (Mg,Fe)2 SiO4 .
Таблица 1
Параметры решетки и разрядные характеристики соединений LiMPO4 [6- 9]
| M | a, Е | b, Е | c, Е | U, В | Емкость, А*час/кг |
| Mn | 10,45 | 6,11 | 4,75 | 4,1 | 140 |
| Fe | 10,31 | 6,00 | 4,69 | 4,3 | 148 |
| Co | 10,20 | 5,92 | 4,68 | 4,8 | 86 |
| Ni | 10,20 | 5,92 | 4,68 |
Фосфаты LiMPO4, где M = Mn, Co, Ni получены в ходе взаимодействия карбоната лития, оксида металла (MO или MnO2 ) и дигидрофосфата аммония - (NH4 )2 HPO4 при температуре 350 °C, которую затем повышали до 780 °C и выдерживали 18 часов на воздухе [6]. LiFePO4 получен аналогично, но в инертной атмосфере [10].
1.3. Смешанные фторидофосфаты щелочных и переходных металлов
Просмотр реферативных журналов, баз данных PDF-2 и ICSD обнаружил только три фазы формульного типа A+ 2 MPO4 F, из них с литием только одна: Li2 NiPO4 F [11]. Известны также Na2 MnPO4 F [12], Na2 MgPO4 F [13], Na4,6 FeP2 O8,6 F0,4 [14, 15, 16, 17].
Структура Li2 NiPO4 F(рис. 1) определена рентгенографически на монокристалле [11]. Она относится к ромбической сингонии (пространственная группа Pnma, параметры a= 10.473(3) Е, b= 6.2887(8) Е, c= 10.846(1) Е, Z=8). В структуре можно выделить рутиловые цепи из октаэдров NiO4 F2 , соединенных ребрами, вытянутые вдоль оси y. Эти цепи соединены в двух остальных измерениях тетраэдрами PO4 . В пустотах каркаса размещаются катионы лития. Половина их находится в уплощенных тетраэдрах из четырех атомов кислорода, четверть – в квадратных пирамидах из 4 O + 1 F и еще одна четверть в сильно асимметричной координации, где трудно сделать однозначный выбор между КЧ 4,5,6. Достаточно короткие (до 3,21 Е) расстояния Li-Li соединяют все позиции лития в двумерную сеть в плоскости y0z (рис. 2). Это позволяет ожидать достаточно высокую подвижность ионов лития в каркасе и возможность их извлечения с окислением никеля и сохранением исходного каркаса:
Li2 Ni2+ PO4 F ® LiNi3+ PO4 F + Li+ + e ® Ni4+ PO4 F + 2 Li+ + 2 e
Но сведений о таких свойствах Li2 NiPO4 F в литературе не обнаружено. Можно было бы ожидать существование аналогичных фаз, содержащих на месте никеля другие катионы близкого размера с переменной степенью окисления (табл. 2), но никаких сведений о них в литературе также не обнаружено.
Таблица 2
Эффективные кристаллохимические радиусы [18] некоторых двухзарядных катионов в октаэдрической координации в высокоспиновом состоянии
| M | Mn2+ | Fe2+ | Co2+ | Ni2+ |
| VI R, Е | 0,97 | 0,92 | 0,885 | 0,83 |
В данной работе поставлена задача получения новых фаз состава Li2 MPO4 F, где M = Mn, Fe, Co, и исследования возможности окислительного извлечения лития из них и из ранее известного никелевого соединения. Предполагалось, что за счет удвоенного содержания лития можно будет повысить емкость электродного материала по сравнению с фазами типа оливина (табл. 3).
Таблица 3
Теоретические удельные емкости некоторых известных и предполагаемых материалов положительного электрода литий-ионного аккумулятора
| Восстановленная форма | Окисленная форма | Емкость, А*час/кг |
| LiMO2 (M = Co, Ni) | Li0.5 MO2 | 140 |
| LiMPO4 (M = Mn, Fe, Co, Ni) | MPO4 | 170 |
| Li2 MPO4 F (M = Mn, Fe, Co, Ni) | LiMPO4 F | 144 |
| MPO4 F | 288 |
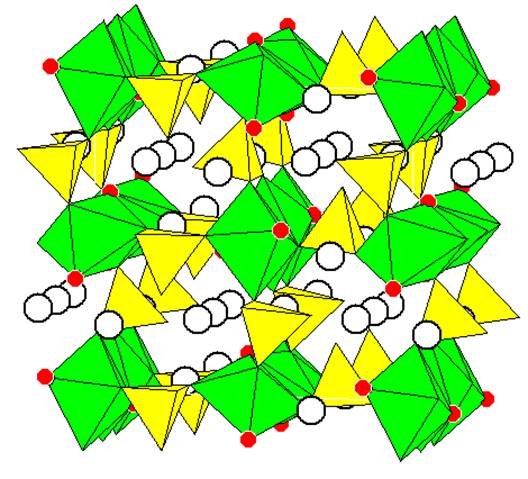 |
Рисунок 1
Полиэдрическое изображение кристаллической структуры Li2 NiPO4 F [10]
Зеленым цветом показаны октаэдры вокруг катионов никеля, желтым – тетраэдры PO4, красным – ионы фтора (в остальных вершинах – кислород), светлыми кружками показаны ионы лития.
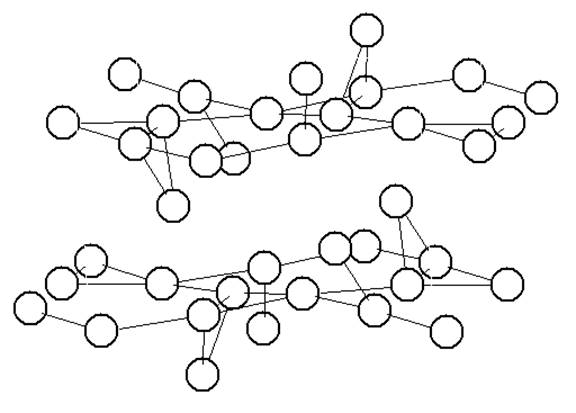 |
Рисунок 2
Система позиций лития в структуре Li2 NiPO4 F. Соединены позиции, отстоящие друг от друга не более чем на 3,21 Е.
2. Исходные вещества и методы экспериментов
2.1. Исходные вещества и их анализ
Фосфор, фтор и литий вводили в виде дигидрофосфата аммония, высушенного при 100 °С, фторида и карбоната лития, высушенных при 200 °С. Реактивный оксид никеля (серый, нестехиометрический) прокаливали при 900 °C для превращения в зеленый стехиометрический NiO. Реактивный оксид кобальта (+2) использовали в непрокаленном виде (рентгенофазовым анализом проверено, что это действительно CoO, а не Co3 O4 ). Для введения переходных металлов испытаны и другие реагенты: карбонаты кобальта и марганца, ацетат никеля, а также оксалаты марганца и железа (+2), осажденные из водных растворов. Для проведения данной части экспериментов брали растворимые соли: сульфат железа (+2) и хлорид марганца (+2), растворяли их в горячей дистиллированной воде и приливали к ним горячий раствор оксалата аммония. После охлаждения осадки отфильтровывали на воронке Бюхнера, промывали дистиллированной водой до удаления сульфат- или хлорид-ионов и высушивали при комнатной температуре несколько дней.
Нет уверенности в том, что эти карбонаты, оксалаты и ацетат точно соответствуют идеальным формулам: при хранении возможны потеря или приобретение воды, гидролиз, окисление. Поэтому потребовалось провести их анализ. Для этого по три параллельных пробы каждого из исходных веществ прокаливали до постоянной массы и взвешивали в виде оксидов. Температуру прокаливания выбирали на основе литературных данных о стабильности весовых форм: для получения Fe2 O3 , NiO – 900 °С, для получения Co3 O4 и Mn2 O3 - 750 °С [19, 20, 21].
2.2. Проведение синтезов
При нагревании фторида лития с дигидрофосфатом аммония возможно улетучивание фтороводорода. Поэтому проведение синтеза в одну стадию вряд ли возможно. Сначала нужно получить LiMPO4 , и лишь после полного удаления воды можно добавлять фторид лития.
Таким образом, можно выделить две стадии.
(1) 2NH4 H2 PO4 +Li2 CO3 + 2MO ® 2 LiMPO4 + 2NH3 + CO2 + 2H2 O.
Здесь MO – это либо оксид (NiO, CoO), либо соединение, разлагающееся до оксида.
(2) LiMPO4 + LiF ® Li2 MPO4 F
Навески веществ смешивали и растирали в яшмовой ступке до полной однородной массы, затем прессовали таблетки, выдерживали при температуре 150-170 °C 2 часа для удаления большей части летучих компонентов (если сразу нагреть до более высоких температур, то происходит оплавление и однородность таблетки нарушается). Затем температуру постепенно повышали, периодически перетирая смесь, до получения практически чистых LiMPO4 . Обжиги проводили либо в муфельной печи, либо в инертной атмосфере в трубчатой печи.
Ввиду отсутствия инертных газов в баллонах, пришлось получать азот нагреванием водного раствора хлорида аммония и нитрита бария. Колба, в которой происходила основная реакция по получению азота (экзотермическая реакция, небольшое нагревание), соединялась с двумя промывалками с сернокислым раствором бихромата калия для улавливания возможных примесей аммиака и оксида азота, далее шла накаливаемая трубка с пористыми медными гранулами для очистки от кислорода и оксидов азота, потом с силикагелем для грубой осушки и две промывалки с концентрированной серной кислотой для более полного улавливания водяных паров. Эти промывалки соединялись с трубкой, в которой находились смеси веществ в спрессованном виде в никелевых лодочках. Вначале через установку пропускали трехкратный объем азота для удаления воздуха и лишь потом начинали нагревание. После завершения обжига образцы охлаждали в токе азота, дабы не допустить окисления воздухом.
Продукты проверяли рентгенофазовым анализом и переходили ко второй стадии экспериментов, для этого полученные таблетки перетирали с рассчитанной навеской фторида лития и, спрессовав, продолжали обжиг либо в муфельной печи, либо в инертной атмосфере в трубчатой печи по уже рассмотренной технологии. Чтобы обеспечить более полное связывание фосфата, фторид лития вводили в пятипроцентном избытке. Этот избыток составляет лишь 0,7 масс. % смеси и менее существенен, чем примесь не прореагировавшего фосфата.
2.3. Рентгенография
Рентгенофазовый анализ производился на дифрактометре ДРОН – 2.0 в медном Кa - излучении. Данное излучение не очень подходит для соединений, в которых присутствуют железо и особенно кобальт, так как оно сильно поглощается атомами этих элементов и возбуждает их собственное рентгеновское излучение. В результате дифракционные максимумы ослабляются, и резко возрастает фон. Поэтому снижается чувствительность фазового анализа, уменьшается число наблюдаемых отражений и ухудшается точность их измерения из-за сильных флуктуаций интенсивности. Чтобы преодолеть эти затруднения, следовало бы использовать рентгеновскую трубку с другим анодом, например, кобальтовым (но тогда бы возникли те же проблемы с соединениями марганца) или установить монохроматор на дифрагированном пучке. Но у нас не было такой возможности, поэтому для уменьшения статистических ошибок съемку кобальтового соединения приходилось повторять по несколько раз.