Реферат: Врожденные и наследственные детские заболевания
Ахондроплазия в общем смысле является нарушением роста костей. Хотя ахондроплазия буквально означает "без формирования хрящей", проблема при заболевании ахондроплазией не в том, что хрящи у больного этой аномалией не формируются, а в преобразовании хрящей в кости, в частности, в длинные кости.
Ахондроплазия является одним из старейших известных врожденных дефектов. Частота заболевания ахондроплазией оценивается в диапазоне от 1 заболевшего на 10000 новорожденных в Латинской Америке до около 12 заболевших на 77000 новорожденных в Дании. Средний показатель по всему миру составляет примерно 1 заболевший на 25000 новорожденных.
Achondroplasia является отличительной условии, что обычно можно отметить при рождении. Ребенок с achondroplasia имеет довольно длинный, узкий торс (ствола) с короткими конечностей (рук и ног), а также непропорционально укорочение проксимального (недалеко от туловища) сегментов конечностей (верхняя оружия и бедер). Существует один, как правило, с большой головой известность в лоб (фронтальный bossing), отсталостью (гипоплазия) в midface с скулы, что отсутствие внимания, и низкий носовой мост с узкими носовых проходов. Ребенка появляются короткие пальцы и Рингера и средний пальцы расходятся дать руку трезубец (три элемента) внешность. Большинство соединений можно распространить более чем нормальным. Например, колени могут hyperextend сверх обычного точки остановки. Не все суставы имеют слабый в этом пути. Напротив, расширение и ротация локте аномально ограничено. Hip продление также, как правило, ограничены.
При рождении там часто известность в середине в нижней части спины с небольшим gibbus (неровности). При ходьбе, неровностей уходит и выраженным влиянием (лордоз) в поясничную область (в нижней части спины), становится очевидной. Поясничного лордоза является стойким. На ноги поклонился (genu Варум).
Младенец экспонатов некоторых снижение мышечного тонуса (гипотония).Из-за большой головой, особенно по сравнению с остальной частью тела, а также уменьшился мышечный тонус, ребенок с achondroplasia будет позади "графика" в достижении обычный двигатель этапов. График которому achondroplastic развития ребенка следует сравнивать не в том, что для всех детей в общей численности населения, а затем расписание achondroplastic детей.
Гемолитическая болезнь новорожденных
Причиной гемолитической болезни новорожденных является несовместимость крови матери и плода по резус-фактору или группе крови. При беременности эритроциты плода попадают в организм матери, вызывая выработку у нее антител против эритроцитов плода. Антитела матери в крови плода приводят к разрушению эритроцитов как до, так и после рождения, стимулируя массивный гемолиз. Незрелая печень ребенка не может вывести из организма продукты распада эритроцитов. Интоксикация приводит к гибели печеночных клеток, клеток подкорки и коры головного мозга (ядерная желтуха). При гемолитической болезни новорожденных выделяют 3 формы течения заболевания: отечную, желтушную и анемическую. Отечная форма встречается сравнительно редко, но протекает особенно тяжело и приводит обычно к внутриутробной гибели плода. Часто наступают преждевременные роды. Родившиеся живыми дети погибают в первые минуты или часы жизни. Желтушная форма проявляется на 1-2-й день жизни ребенка и характеризуется желтухой, гепатоспленомегалией, анемией. Отмечается пастозность тканей. Дети адинамичны, плохо сосут, рефлексы снижены. Билирубиновая интоксикация может достигать степени ядерной желтухи. Улучшение состояния можно наблюдать спустя 2-3 недели, но прогноз для дальнейшего развития ребенка неблагоприятен. Самой легкой является анемическая форма. Анемия развивается на первой или второй неделе жизни. Характерны бледность, плохой аппетит, вялость, гепатоспленомегалия, уровень билирубина повышен умеренно.
Микроцефалия
Микроцефалия - значительное уменьшение размеров черепа и мозга, сопровождающееся задержкой психического развития и различными неврологическими нарушениями. Принято выделять первичную и вторичную микроцефалию. Первичная - наследственное заболевание с рецессивным типом передачи от родителей потомству. При этой форме уже у новорожденного размеры черепа значительно уменьшены, а масса мозга снижена до 250 - 300 г (в норме - около 400 г). Вторичная микроцефалия развивается в результате действия на мозг различных вредностей (гипоксия, инфекции, травма, нарушения обмена веществ). В головном мозге, помимо резкого уменьшения массы, находят очаги деструкции, следы воспалительного процесса, кровоизлияния и т. д. Желудочковая система мозга расширена. У доношенного новорожденного с микроцефалией окружность черепа составляет не более 25 - 27 см. Роднички отсутствуют, кости черепа плотные. Лицевая часть черепа преобладает над мозговой. Характерен «убегающий» назад лоб, выступающие надбровные дуги, низко расположенные большие оттопыренные уши, высокое и узкое небо. В некоторых случаях симптомы микроцефалии формируются в первые месяцы жизни. Окружность черепа не увеличивается или увеличивается значительно меньше нормы, быстро смыкаются швы черепа, закрываются роднички. Из неврологических нарушений встречаются изменения мышечного тонуса, судороги, тремор, нарушения координации движений, параличи или парезы, косоглазие, задержка развития статических и двигательных функций. Неврологические нарушения более выражены у детей с вторичной микроцефалией. Типичным признаком является интеллектуальный дефект - от глубокой имбицильности до идиотики. При всех микроцефалиях страдают развитие речи (она может полностью отсутствовать) и эмоциональная сфера. Прогноз зависит от степени недоразвития мозга. В социальной адаптации больных большое значение придается воспитанию и обучению, при которых используются сохранные эмоции, механическая память и способность к подражанию
Врожденные пороки сердца
Врожденными называют те заболевания , которые развились еще до рождения или во время родов. Многие виды врожденной патологии сердца и кровеносных сосудов встречаются не только порознь, но и в различных сочетаниях примерно у 1 из каждых 200 новорожденных.
Частота врожденных пороков сердца достаточна высока. У разных авторов оценка частоты встречаемости колеблется, но, в среднем, она составляет 0,8 - 1,2% от всех новорожденных. Из числа всех встречающихся пороков развития она составляет до 30%.
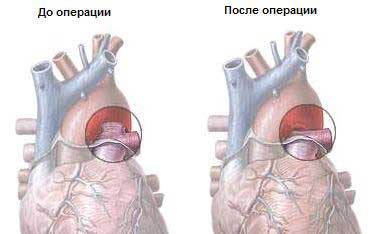
Причины большинства врожденных пороков сердечно-сосудистой системы остаются неизвестными. При наличии в семье одного ребенка с пороком сердца риск рождения других детей с такого рода пороком несколько возрастает, но все же остается низким: от 1 до 5%. Врожденные пороки сердца (ВПС) представляют собой весьма обширную и разнородную группу заболеваний, в которую входят как относительно легкие формы, так и состояния, несовместимые с жизнью. Основная масса детей погибает в течении первого года жизни (до 70-90%), а из них в течении первого месяца. После первого года жизни смертность резко снижается, и в период от 1 года до 15 лет погибают не более 5% детей. Понятно, что это большая и серьезная проблема. Мы с вами рассмотрим только некоторые ее аспекты.
Предрасполагающие факторы
Прежде всего, какие причины могут приводить к рождению ребенка с ВПС.
Некоторыми исследованиями показано, что существует определенная сезонность в рождении детей с ВПС. Например, есть данные, что открытый артериальный проток встречается преимущественно у девочек, родившихся во второй половине года, чаще всего с октября по январь. Мальчики с коарктацией аорты чаще рождаются в марте и апреле, наиболее редко - в сентябре и октябре.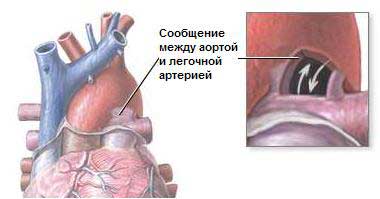
Безусловно, одного лишь наличия вирусного заболевания еще недостаточно для того, чтобы у будущего ребенка развился порок сердца, однако при условии дополнительных факторов (тяжести вирусного, да и бактериального заболевания, наличия генетической предрасположенности к неблагоприятным реакциям на пусковое воздействие данного фактора) вирусный агент может оказаться решающим в плане формирования врожденного порока сердца у новорожденного.
Генетической предрасположенности
Несомненным фактором риска является наличие генетической предрасположенности. Чаще всего при объяснении типа наследования прибегают к так называемой полигенно- мультифакториальной модели. По этой модели, чем более тяжелый порок сердца есть в семье, тем выше риск его повторения, чем больше родственников страдают ВПС, тем выше риск повторного поражения и т.д. Помимо подобного типа наследования, есть еще и генные мутации и хромосомные аномалии. Дать точную количественную оценку риска рождения ребенка с ВПС может (да и то не всегда) только генетик в процессе проведении медико-генетического консультирования. Факторами риска рождения ребенка с ВПС таким образом являются: возраст матери, эндокринные нарушения у супругов, токсикозы в I триместре и угрозы прерывания беременности, мертворождения в анамнезе, наличие других детей с врожденными пороками развития, прием женщиной эндокринных препаратов для сохранения беременности и др.
Классификация врожденных пороков сердца
Предложено несколько классификаций врожденных пороков сердца, общим для которых является принцип подразделения пороков по их влиянию на гемодинамику. Наиболее обобщающая систематизация пороков характеризуется объединением их, в основном по влиянию на легочный кровоток, в следующие 4 группы.
- I. Пороки с неизмененным (или мало измененным) легочным кровотоком: аномалии расположения сердца, аномалии дуги аорты, ее коарктация взрослого типа, стеноз аорты, атрезия аортального клапана; недостаточность клапана легочного ствола; митральные стеноз, атрезия и недостаточность клапана; трехпредсердное сердце, пороки венечных артерий и проводящей системы сердца.
- II. Пороки с гиперволемией малого круга кровообращения:
1 )не сопровождающиеся ранним цианозом - открытый артериальный проток, дефекты межпредсердной и межжелудоч-ковой перегородок, синдром Лютамбаше, аортолегочный свищ, коарктация аорты детского типа
2) сопровождающиеся цианозом -трикуспидальная атрезия с большим дефектом межжелудочковой перегородки, открытый артериальный проток с выраженной легочной гипертензией и током крови из легочного ствола в аорту
- III. Пороки с гиповолемией малого круга кровообращения:
1) не сопровождающиеся цианозом - изолированный стеноз легочного ствола
2) сопровождающиеся цианозом - триада, тетрада и пентада Фалло, трикуспидальная атрезия с сужением легочного ствола или малым дефектом межжелудочковой перегородки, аномалия Эбштейна (смещение створок трикуспидального клапана в правый желудочек), гипоплазия правого желудочка
- IV. Комбинированные пороки с нарушением взаимоотношений между различными отделами сердца и крупными сосудами: транспозиция аорты и легочного ствола (полная и корригированная), их отхождение от одного из желудочков, синдром Тауссиг - Бинга, общий артериальный ствол, трехкамерное сердце с единым желудочком и др.
Приведенное подразделение пороков имеет практическое значение для их клинической и особенно рентгенологической диагностики, т. к. отсутствие или наличие изменений гемодинамики в малом круге кровоoбращения и их характер позволяют отнести порок к одной из групп I-III или предположить пороки IV группы, для диагностики которых необходима, как правило, ангиокардиография.
Некоторые врожденные пороки сердца (особенно IV группы) встречаются весьма редко и только у детей. У взрослых из пороков 1-II групп чаще выявляются аномалии расположения сердца (прежде всего декстрокардия), аномалии дуги аорты, ее коарктация, аортальный стеноз, открытый артериальный проток, дефекты межпредсердной и межжелудочковой перегородок; из пороков III группы - изолированный стеноз легочного ствола, триада и тетрада Фалло.
Клинические проявления и течение определяются видом порока, характером гемодинамических нарушений и сроками наступления декомпенсации кровообращения.
Пороки, сопровождающиеся ранним цианозом (так наз. "синие" пороки), проявляются сразу или вскоре после рождения ребенка. Многие пороки, особенно I и II группы, долгие годы имеют бессимптомное течение, выявляются случайно при профилактическом медицинском обследовании ребенка или при появлении первых клинических признаков нарушений гемодинамики уже в зрелом возрасте больного.
Пороки III и IV групп могут относительно рано осложняться сердечной недостаточностью, приводящей к летальному исходу.
Врожденные заболевания.
Альбинизм
Альбинизм — это врожденное нарушение, характеризующееся недостаточной окраской кожи, волос, глаз. Это заболевание существует с рождения, и причиной его является недостаток или отсутствие пигмента (красящего вещества). Альбинизм — явление редкое, всречающееся у одного из 10 000 новорожденных в мире. Это нарушение отмечается у разных народов в разных частях света. Различают шесть типов альбинизма. Наиболее часто встречается полный альбинизм, характеризующийся молочно-белой окраской волос, тонкой и шелковистой их структурой, белыми ресницами и бровями, белой или нежно-розовой кожей и красными или розовыми глазами (которые в зависимости от освещения могут казаться светло-серыми или голубыми). Другие виды альбинизма встречаются реже и характеризуются более сильной окраской кожи, волос, глаз.
Альбинизм проявляется лишь в том случае, если оба родителя, являясь носителями рецессивных дефектных генов, передадут их ребенку. Если у пары с одинаковым типом альбинизма рождаются дети, то все они будут иметь тот же тип альбинизма. Альбинизм связан с близкородственными браками; исследованиями доказана высокая частота наличия общих предков у родителей детей с альбинизмом. Однако у пары с разными типами альбинизма есть шанс рождения здорового ребенка. У всех детей, унаследовавших два дефектных гена, отмечается снижение или полное отсутствие тирозиназы (фермента, необходимого для нормальной окраски кожи, волос, глаз).
Кроме уже упомянутых особенностей окраски кожи, волос, глаз, некоторые люди с альбинизмом бывают низкорослы. При полном альбинизме глаза имеют повышенную чувствительность к свету, свет раздражает их, движения глаз быстрые. Часто отмечается астигматизм (нарушение зрения, при котором ребенок с трудом концентрирует зрение на горизонтальных и вертикальных объектах одновременно; см. нарушения зрения).
Альбинизм как диагноз ставится на основании осмотра при наличии у ребенка характерной окраски кожи, волос, глаз. Диагноз может быть дополнен лабораторными исследованиями, подтверждающими отсутствие тирозиназы. Альбинизм не может быть выявлен до родов при проколе плодного пузыря у матери.
Альбинизм часто приводит к нарушениям зрения вследствие повышенной чувствительности к свету. При повреждении кожи солнечным светом нормальной или высокой интенсивности могут возникать тяжелый ожог и рак кожи. Жертвы альбинизма обычно страдают бесплодием и умирают раньше, чем здоровые.
Лечения альбинизма не существует. Однако люди с альбинизмом могут максимально адаптироваться в жизни, если будут соблюдать следующие предосторожности: очень важно, чтобы дети с альбинизмом были максимально защищены от воздействия солнечного света. На открытом воздухе они должны носить одежду, полностью закрывающую тело, пользоваться солнцезащитными кремами или лосьонами даже в облачную погоду. Если кожа подвергается влаге, солнцезащитный крем следует нанести повторно.
Перед поступлением в школу ребенка с альбинизмом следует показать офтальмологу для того, чтобы подобрать ему защитные (темные) очки, или, при нарушении зрения, корригирующие очки или контактные линзы.
Родители, у которых в семье раньше отмечался альбинизм , должны обратиться в генетическую консультацию, чтобы попытаться определить возможность генетического заболевания на основании родословной.
Гемофилия
Гемофилия - наследственная болезнь, передаваемая по рецессивному сцепленному с Х-хромосомой типу, проявляющаяся повышенной кровоточивостью.
Этиология и патогенез . Передается по наследству через потомство сестер и дочерей больного. Женщины-кондукторы передают гемофилию не только своим детям, а через дочерей-кондукторов — внукам и правнукам, иногда и более позднему потомству. Болеют мальчики (гемофилия С встречается и у девочек). Выделяют три формы гемофилии —А, В и С. При гемофилии А отсутствует фактор VIII, при гемофилии В — фактор IX и при гемофилии С — фактор XI свертывания крови.
Первые проявления кровоточивости у больных гемофилией развиваются чаще всего в то время, когда ребенок начинает ходить или подвергается бытовым травмам. У некоторых больных первые признаки гемофилии выявляются уже в период новорожденности (кефалгематома, синяки на теле, подкожные гематомы). Гемофилия может проявиться и в грудном возрасте, но угрожающих жизни кровотечений обычно не бывает. Это можно объяснить тем, что в женском молоке содер корригирует дефект крови больных гемофилией. Дети, страдающие гемофилией, отличаются хрупкостью, бледной тонкой кожей и слабо развитым подкожным жировым слоем. Кровотечения по сравнению с вызвавшей их причиной всегда бывают чрезмерными. Наряду с подкожными, внутримышечными, межмышечными наблюдаются кровоизлияния во внутренние органы, а также гемартрозы, протекающие с повышением температуры. Чаще всего поражаются крупные суставы. Повторные кровоизлияния в один и тот же сустав ведут к воспалительным изменениям его, деформации и анкилозу.
Диагноз основывается на генеалогическом анализе, выявлении резкого замедления свертываемости крови. Симптомы Кончаловского, Румпеля—Лееде, Коха отрицательные. Ретракция кровяного сгустка нормальная или несколько замедлена. Снижен уровень факторов VIII и IX. Для определения формы гемофилии предложен тест генерации тромбопластина.
При дифференциальном диагнозе следует иметь в виду апластическую анемию, хронические формы лейкоза, полицитемию, тяжелые септические заболевания и другие формы геморрагического диатеза .
Раны следует очистить от сгустков и промыть раствором пенициллина в изотоническом растворе натрия хлорида. Затем накладывают марлю, пропитанную одним из кровоостанавливающих (адреналин, перекись водорода и др.) и богатых тромбопластином средств (гемостатические губки, грудное молоко). При гемофилии в качестве кровоостанавливающего средства может использоваться сыворотка крови человека и животных. Кровоточащая рана должна быть хорошо затампонирована.
При гемофилии А следует переливать свежую кровь, так как при хранении в консервированной крови быстро инактивируется антигемофильный глобулин А. При гемофилии В можно переливать обычную донорскую кровь, ибо она содержит достаточное количество компонента тромбопластина плазмы. Для остановки кровотечения обычно достаточно переливания малых доз крови (30—50 мл). При значительных кровопотерях применяют вливания больших доз крови (для младших детей по 5 мл на 1кг массы тела, для старших — однократная доза 150—200 мл).
В последнее время при гемофилии А в/в вводят антигемофильную плазму (разовая доза 50—100 мл), антигемофильную плазму с эпсилон-аминокапроновой кислотой, сухую антигемофильную плазму (разводят дистиллированной водой — 100:50 мл). Кроме того, используют антигемофильный глобулин (разовая доза 5 мл в/в); при гемофилии В и С с хорошим эффектом применяют человеческую сыворотку (20 мл) и эпсилон-аминокапроновую кислоту (5% раствор до 100 мл старшим детям 3—4 раза в сутки).
Прогноз при современном лечении благоприятный.
Профилактика — медико-генетическое консультирование.
Синдром Дауна
Синдром Дауна - одно из самых распространенных генетических нарушений. Частота рождения детей с синдромом Дауна составляет примерно один на 600-800 новорожденных.В нашей стране чаще всего используется термин "болезнь Дауна". Причем нередко говорится, что это "неизлечимая болезнь". Некоторые специалисты утверждают, что существует даже два диагноза: болезнь Дауна и синдром Дауна. Они уверяют, что состояние ребенка зависит от того, имеется ли у него болезнь или синдром. Подобные утверждения являются крайне некорректными и даже абсурдными. Синдром Дауна не является болезнью . Слово "синдром" означает определенный набор признаков, или особенностей. Впервые признаки людей с синдромом Дауна описал английский врач Джон Лэнгдон Даун (Down) в 1866 году. Его имя и послужило названием для данного синдрома - синдрома Дауна. Однако лишь в 1959 году французский ученый Жером Лежен (Lejeune) обнаружил причину синдрома. Причиной, которая вызывает синдром Дауна, является лишняя хромосома. Каждая клетка человеческого тела обычно содержит 46 хромосом. Хромосомы несут в себе признаки, которые наследует человек от родителей, и расположены они парами - половина от матери, половина от отца. У людей с синдромом Дауна в 21-й паре присутствует дополнительная хромосома, то есть имеет место так называемая трисомия, поэтому в клетках организма оказывается по 47 хромосом. Диагноз "синдром Дауна" может быть поставлен только врачом-генетиком с помощью анализа крови, показывающего наличие лишней хромосомы. Дополнительная хромосома появляется в результате случайности при образовании яйцеклетки или сперматозоида, либо во время первого деления клетки, которое следует за оплодотворением (то есть когда яйцеклетка и сперматозоид сливаются). До сих пор не сложилось однозначного мнения о том, что служит причиной такой генетической аномалии. Дети с синдромом Дауна рождаются с одинаковой частотой во всех странах мира, независимо от уровня благосостояния или экологии. Такие дети рождаются в семьях академиков и строителей, президентов и безработных. Появление ребенка с синдромом Дауна не зависит от образа жизни, национальности, уровня образования или социального положения родителей. Ничьей "вины" в появлении на свет такого ребенка нет.
Чем отличается малыш с синдромом Дауна от других детей?
Наличие этой дополнительной хромосомы обусловливает появление ряда физиологических особенностей, вследствие которых ребенок будет медленнее, чем его ровесники, развиваться и проходить общие для всех детей этапы развития. Раньше считалось, что все люди с синдромом Дауна имеют тяжелую степень умственной отсталости и не поддаются обучению. Современные исследования показывают, что практически все люди с синдромом Дауна отстают в интеллектуальном развитии, но внутри этой группы их интеллектуальный уровень сильно различается - от незначительного отставания до средней и тяжелой степени отставания. Все-таки большинство детей с синдромом Дауна могут научиться ходить, говорить, читать, писать и, вообще, делать большую часть того, что умеют делать другие дети, нужно лишь обеспечить им адекватную среду жизни и соответствующие программы обучения. В нашей стране представления о людях с синдромом Дауна носят, скорее, мифический, чем реальный характер, и отношение к ним часто противоположно. Одни уверяют, что люди с синдромом Дауна всегда агрессивны, часто сексуально агрессивны, при этом их агрессия может быть направлена даже на близких членов семьи, так как они настолько глупы, что не способны отличить, например, своих родителей от других людей, их невозможно чему-либо научить и их поведение, даже во взрослом возрасте, всегда будет неадекватным. Другое мнение полностью противоположное: люди с синдромом Дауна всегда очень добрые, веселые, общительные, они очень музыкальны, любят петь и танцевать и даже могут стать танцорами и музыкантами, несмотря на то, что уровень интеллекта у них крайне низкий.
Правды мало как в первом, так и во втором утверждении. Люди с синдромом Дауна , несмотря на внешнюю схожесть, отличаются друг от друга так же, как и люди без синдрома Дауна . У разных людей с синдромом Дауна разные умственные способности, разное поведение и физическое развитие. Каждый человек обладает уникальной личностью, индивидуальностью, способностями и талантами. Они во многом похожи на своих родителей, и, как и у любого другого человека, у каждого из них свой характер и темперамент. Люди с синдромом Дауна могут гораздо лучше развить свои способности, если они живут дома, в атмосфере любви, если в детстве они занимаются по программам ранней помощи, если они получают специальное образование, надлежащее медицинское обслуживание и ощущают позитивное отношение к себе общества.
Ребенок-олигофрен. "Неизлечим" не значит "обречен"