Контрольная работа: Естествознание на молекулярном уровне
Орг. химия изучает не только соединения, получаемые из растительных и животных организмов, но в основном соединения, созданные искусственно с помощью лаборатории или промышленного органического синтеза. Более того, объектами изучения компьютерной орг. химии являются соединения, не только не существующие в живых организмах, но которые, по-видимому, нельзя получить искусственно (напр., гипотетический аналог метана, имеющий не природное тетраэдрич. строение, а форму плоского квадрата, в центре которого лежит атом С, а в вершинах – атомы Н).
Классификация органических соединений. Основу орг. соединений составляет незамкнутая (открытая) или замкнутая цепь углеродных атомов; одно или несколько звеньев цепи может быть заменено на атомы, отличные от углерода, – гетероатомы, чаще всего О, N, S. По структуре орг. соед. подразделяют на алифатические соединения – углеводороды и их производные, имеющие открытую углеродную цепь; карбоциклические соединения с замкнутой углеродной цепью; гетероциклические соединения. Углеводороды и их производные, не содержащие кратных связей, относятся к насыщенным соединениям, с кратными связями – к ненасыщенным.
Историческая справка . Истоки орг. химии восходят к глубокой древности (уже тогда знали о спиртовом и уксуснокислом брожении, крашении индиго и ализарином). Однако в средние века (период алхимии) были известны лишь немногие индивидуальные орг. вещества. Все исследования этого периода сводились главным образом к операциям, при помощи которых, как тогда думали, одни простые вещества можно превратить в другие. Начиная с ХVI в. (период ятрохимии) исследования были направлены в основном на выделение и использование различных лекарственных веществ: был выделен из растений ряд эфирных масел, приготовлен диэтиловый эфир, сухой перегонкой древесины получены древесный (метиловый) спирт и уксусная кислота, из винного камня – винная кислота, перегонкой свинцового сахара – уксусная кислота, перегонкой янтаря – янтарная.
Слияние хим. соединений растительного и животного происхождения в единую хим. науку орг. химии осуществил Й. Берцелиус, который ввел сам термин и понятие орг. вещества, образование последнего, по Берцелиусу, возможно только в живом организме при наличии «жизненной силы». Это заблуждение опровергли Ф. Вёлер (1828), который получил мочевину (орг. вещество) из цианата аммония (неорг. вещество), А. Кольбе, синтезировавший уксусную кислоту, М. Бертло, получивший метан из H2 S и CS2 , A.M. Бутлеров, синтезировавший сахаристые вещества из формалина. В первой пол. XIX в. был накоплен обширный опытный материал и сделаны первые обобщения, определившие бурное развитие орг. химии: развиты методы анализа орг. соединения (Берцелиус, Ю. Либих, Ж. Дюма, М. Шеврёль), создана теория радикалов (Вёлер, Ж. Гей-Люссак, Либих, Дюма) как групп атомов, переходящих неизменными из исходной молекулы в конечную в процессе реакции; теория типов (Ш. Жерар, 1853), в которой орг. соединения конструировались из неорг. веществ – «типов» замещением в них атомов на орг. фрагменты; введено понятие изомерии (Берцелиус).
Одновременно продолжается интенсивное развитие синтеза. Создаются первые промышленные производства орг. соед. (А. Гофман, У. Перкин-старший – синтетич. красители: мовеин, фуксин, цианиновые и азокрасители). Усовершенствование открытого Н.Н. Зининым (1842) способа синтеза анилина послужило основой создания анилинокрасочной промышленности.
Идея неразрывной связи хим. и физ. свойств молекулы с ее строением, идея единственности этого строения впервые была высказана Бутлеровым (1861), который создал классическую теорию хим. строения (атомы в молекулах соединяются согласно их валентностям, хим. и физ. свойства соединения определяются природой и числом входящих в их состав атомов, а также типом связей и взаимным влиянием непосредственно несвязанных атомов). Теория хим. строения определила дальнейшее бурное развитие орг. химии: в 1865 Кекуле предложил формулу бензола, позднее высказал идею об осцилляции связей; В.В. Марковников и А.М. Зайцев сформулировали ряд правил, впервые связавших направление хим. реакции с хим. строением вступающего в реакцию вещества.
Работами Байера, К. Лаара, Л. Клайзена, Л. Кнорра развиты представления о таутомерии – подвижной изомерии. Все эти теоретические представления способствовали мощному развитию синтетической химии. К кон. XIX в. были получены все важнейшие представители углеводородов, спиртов, альдегидов и кетонов, карбоновых кислот, галогено- и нитропроизводных, азот- и серосодержащих структур, гетероциклов ароматической природы. Разработаны методы получения диенов, ацетиленов и алленов (А.Е. Фаворский). Открыты многочисленные реакции конденсации (Ш. Вюрц, А.П. Бородин, У. Перкин, Клайзен, А. Михаэль, Ш. Фридель, Дж. Крафтс, Э. Кнёвенагель и др.). Исключительные успехи были достигнуты Э.Г. Фишером в изучении углеводов, белков и пуринов, в использовании ферментов в орг. синтезе (1894), им же был осуществлен синтез полипептидов. Основой промышленности душистых веществ становятся работы О. Валлаха по химии терпенов. Выдающимися даже для нашего времени являются пионерские работы Р. Вильштеттера. Фундаментальный вклад в развитие орг. синтеза был внесен В. Гриньяром (1900–20) и Н.Д. Зелинским (1910) – создание исключительно плодотворного метода синтеза магнийорганических соединений и открытие каталитических превращений углеводородов; последнее сыграло выдающуюся роль в развитии химии нефти. Химия свободных радикалов началась с работ М. Гомберга (1900), открывшим трифенилметильный радикал, и была продолжена работами А.Е. Чичибабина, Г. Виланда и Ш. Гольдшмидта.
Строение органических соединений. Для орг. соединений характерны неполярные ковалентные связи С–С и полярные ковалентные связи С–О, С–N, С–Hal, С–металл и т.д. Образование ковалентных связей было объяснено на основании развитых Г. Льюисом и В. Косселем (1916) предположений о важной роли электронных образований – октетов и дублетов. Молекула устойчива, если валентная оболочка таких элементов, как С, N, О, Hal, содержит 8 электронов (правило октета), а валентная оболочка водорода – 2 электрона. Хим. связь образуется обобществленной парой электронов различных атомов (простая связь). Двойные и тройные связи образуются соотвующимися двумя и тремя такими парами. Электроотрицательные атомы (F, О, N) используют для связи с углеродом не все свои валентные электроны; «неиспользованные» электроны образуют неподеленные (свободные) электронные пары. Полярность и поляризуемость ковалентных связей в орг. соединениях в электронной теории Льюиса – Косселя объясняется смещением электронных пар от менее электроотрицательного к более электроотрицательному атому, что находит выражение в индуктивном эффекте и мезомерном эффекте.
Классическая теория хим. строения и первоначально электронные представления оказались не в состоянии удовлетворительно описать на языке структурных формул строение многих соединений, например, ароматических. Современная теория связи в орг. соединениях основана главным образом на понятии орбиталей и использует методы молекулярных орбиталей. Интенсивно развиваются квантовохим. методы, объективность которых определяется тем, что в их основе лежит аппарат квантовой механики, единственно пригодный для изучения явлений микромира.
Общая характеристика реакций органических соединений. Реакции орг. соединений имеют некоторые специфические особенности. В реакциях неорг. соединений обычно участвуют ионы; эти реакции протекают очень быстро, иногда мгновенно при нормальной температуре. В реакциях орг. соединений обычно участвуют молекулы; при этом одни ковалентные связи разрываются, а другие образуются. Такие реакции протекают медленнее ионных, и для их ускорения часто требуется повысить температуру или добавить катализатор. Наиболее часто используют в качестве катализаторов и основания. Обычно протекает не одна, а несколько реакций, так что выход нужного продукта очень часто составляет менее 50%.
Возникновение органических соединений. Большинство орг. соединений в природе образуется в процессе фотосинтеза из диоксида углерода и воды под действием солнечного излучения, поглощаемого хлорофиллом в зеленых растениях. Однако орг. соединений должны были существовать на земле и до возникновения жизни, которая не могла появиться без них. Первичная земная атмосфера около 2 млрд. лет назад имела восстановительные свойства, т.к. в ней не было кислорода, а содержались прежде всего водород и вода, а также СО, азот, аммиак и метан.
В условиях сильного радиоактивного излучения земных минералов и интенсивных атмосферных разрядов в атмосфере протекал абиотический синтез аминокислот по схеме: CH4 + H2 O + NH3 ![]() Аминокислоты.
Аминокислоты.
Возможность такой реакции в настоящее время доказана лабораторными опытами.
3. Реакционная способность веществ, анализ и синтез
3.1 Реакционная способность веществ
Реакционная способность, характеристика относительной хим. активности молекул, атомов, ионов, радикалов. Для количественной оценки рассматривают реакционные серии, т.е. ряды однотипных реакций, проводимых в одинаковых условиях, например: (стандартная реакция)
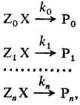
где Х – общая группа атомов, которая претерпевает изменения в данной реакции (реакционный центр), Z0 , Zl ,…, Zn – неизменяющиеся молекулярные фрагменты, Р0 , Р1 ,…, Рn – продукты реакции. Отношения констант скоростей k1 /k0 ,…,kn /k0 количественно характеризуют реакционную способность. В ряду реагентов Zi X(i = 0, 1,…, п). В правильно составленной реакции серии изменение механизма реакции должно быть исключено, т.е. константы скорости должны характеризовать одну и ту же элементарную реакцию.
Типичные реакционные серии. Простейшая ситуация возникает при анализе изомерного состава продуктов реакции. В реакции замещения в ароматическом ряду в зависимости от заместителя R образуются те или иные изомеры, например, при нитровании:
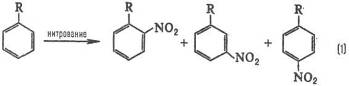
Электронодонорные заместители [R=СН3 , ОСН3 , N(CH3 )2 ] стимулируют образование орто- и пара-продуктов, а электроноакцепторные (R = СООН, SO3 H, NO2 ) – мета-продуктов, причем в первом случае реакция идет легче, чем с незамещенным бензолом (R = Н), а во втором – труднее. Эти закономерности называются правилами ориентации в ароматическом ряду. Стереохимическая направленность перипиклической реакций определяется Вудворда-Хофмаина правилами, например:
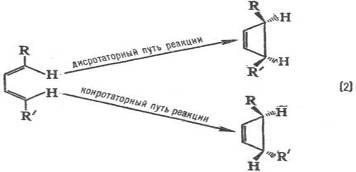
При дисротаторном пути реакции заместители R и R' в продукте будут расположены по одну сторону плоскости цикла, при конротаторном пути – по разные стороны. Эксперимент показывает, что термическая циклизация производных бутадиена происходит по конротаторному пути, а фотохимическая циклизация – по дисротаторному пути.
В примерах (1) и (2) нет необходимости в количественных кинетических измерениях, реакционная способность определяется по относительному выходу изомеров. Пример широкой реакционной серии – реакции радикального присоединения по двойной связи:
![]()
Реакционная способность характеризуется отношением константы скорости k к константе скорости k0 реакции с этиленом (R, R' = Н) (см. табл.). Аналогичные кинетические измерения сделаны для реакций присоединения метильного радикала к ароматическим молекулам и для реакций присоединения др. радикалов.

Квантовохимическая теория реакционной способности. Современная теоретическая химия позволяет непосредственно рассчитать константы скорости только для несложных хим. систем. В теории реакционной способности качественной закономерности могут быть выявлены для объектов любой сложности. При этом используют различные подходы. При эмпирическом подходе классифицируют влияние заместителей по нескольким типам (эффекты сопряжения, полярные, пространственные и др.) и применяют корреляционные соотношения. Традиционный квантовохимический подход основан на активированного комплекса теории; при этом предполагается, что для всех реакций, составляющих реакционную серию, остается примерно постоянным множитель А в Аррениуса уравнении для константы скорости k=Aexp(-E. /RT). Поэтому характеристикой реакционной способности служит энергия активации реакции E. , которая практически совпадает с высотой потенциального барьера на поверхности потенциальной энергии (ППЭ).
Советскому ученому Н.Н. Семенову предстояло открыть разветвленные цепные реакции . Теория разветвленных цепных реакций дала начало новому направлению исследований – химической физике, дисциплине, промежуточной между физикой и химией.
В химии были также открыты колебательные реакции, получившие название «химических часов». Основа колебательной реакции – наличие двух типов молекул, способных превращаться друг в друга. Назовем один из них А (красные молекулы), другой – В (синие). Мы привыкли думать, что химическая реакция – это хаотические, происходящие наобум столкновения частиц. По этой логике взаимные превращения А и В должны приводить к усредненному цвету раствора со случайными вспышками красного и синего. Но когда условия далеки от равновесных, происходит совершенно иное: раствор в целом становится красным, потом синим, потом снова красным. Получается, будто молекулы как бы устанавливают связь между собой на больших, макроскопических расстояниях через большие, макроскопические отрезки времени. Появляется нечто похожее на сигнал, по которому все А или все В реагируют разом… Такое поведение традиционно приписывалось только живому – теперь же ясно, что оно возможно и у систем сравнительно простых, неживых.
3.2 Анализ
Молекулярный анализ, установление качественного и количественного состава хим. соединений и их смесей.