Реферат: Исследование растворимости и ионного обмена как инструмент изучения равновесий в водном растворе
Измерение растворимости труднорастворимых твердых веществ в водных растворах комплексообразугощего агента — один из самых старых методов изучения равновесия в растворе. В конце прошлого столетия этим способом были исследованы молекулярные комплексы пикриновой кислоты, а несколько лет позднее прямой метод и метод конкурирующей растворимости были использованы для определения констант устойчивости комплексов ионов металлов. Этот метод был также применен для изучения равновесия в смешанных водно-органических растворителях и в системах, насыщенных по отношению к труднорастворимым жидкостям или газам.
Большинство данных по растворимости трудно интерпретировать, так как часто происходят значительные изменения состава водной фазы и, следовательно, стехиометрического произведения растворимости
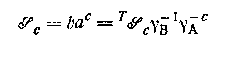
труднорастворимого комплекса ВАс; здесь T ![]() — термодинамическое произведение растворимости. Однако так же, как и в работах с гомогенными системами, для изучения растворимости можно использовать постоянную ионную среду; по методикам Эдмондса и Бирнбаумя, Кинга, Ледена и сотрудников был проведен ряд измерений растворимости с помощью растворов, в которых контролировались коэффициенты активности. Например, Нильссон нашел, что произведение растворимости иодида таллия было одинаково в 4 М растворе перхлората натрия и 4 М растворе иодида натрия. Однако значения
— термодинамическое произведение растворимости. Однако так же, как и в работах с гомогенными системами, для изучения растворимости можно использовать постоянную ионную среду; по методикам Эдмондса и Бирнбаумя, Кинга, Ледена и сотрудников был проведен ряд измерений растворимости с помощью растворов, в которых контролировались коэффициенты активности. Например, Нильссон нашел, что произведение растворимости иодида таллия было одинаково в 4 М растворе перхлората натрия и 4 М растворе иодида натрия. Однако значения ![]() для хлорида, бромида и тиоцианата таллия увеличивались с концентрацией свободного лиганда в области 0,5 М≤а≤4,0 М в 4 М ионной среде, указывая на то, что коэффициенты активности зависят от состава среды при этих условиях.
для хлорида, бромида и тиоцианата таллия увеличивались с концентрацией свободного лиганда в области 0,5 М≤а≤4,0 М в 4 М ионной среде, указывая на то, что коэффициенты активности зависят от состава среды при этих условиях.
Экспериментальные методы
Методы для определения растворимости в широких пределах экспериментальных условий были рассмотрены Циммерманом. При изучении равновесия в водном растворе изменение растворимости в зависимости от начального состава водной фазы измеряется при постоянной температуре.
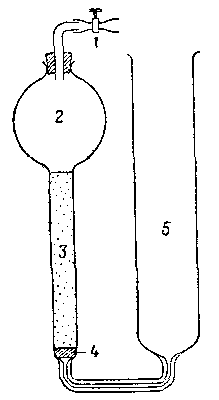
Рис. 1. Сатуратор Бренстеда — Дэписа.
1 – винтовой зажим, 2 – емкость, содержащая растворитель, 3 - колонка с тонкоразмельченным насыщающим твердым веществом; 4— пористая стеклянная прокладка; 5 - емкость, содержащая насыщенный раствор.
Равновесие между твердой фазой и раствором может быть достигнуто или встряхиванием в закрытом сосуде, или пропусканием водной фазы через сатуратор, наполненный твердым веществом; тип сатуратора, примененный Дэвисом и сотрудниками, показан на рис. 1. Равновесие следует устанавливать в термостате. Так как часто равновесие достигается медленно, следует проверять, получается ли одинаковое значение растворимости при ненасыщенной или пересыщенной начальной водной фазе, а также через различные промежутки времени. Равновесие в инертных системах [например, аммиакаты кобальта(Ш)] достигается быстрее с помощью катализатора. Иногда необходимо покрывать внутреннюю поверхность сосудов и пробок парафиновым воском или силиконом для того, чтобы избежать потерь растворенного вещества вследствие сорбции на стекле.
Твердая фаза может быть отделена от насыщенного раствора фильтрованием или центрифугированием, которые следует проводить при температуре равновесия. Особенно удобно помещение с постоянной температурой, но, если его нет, фильтрование можно легко выполнить в термостате. Например, установление равновесия и последующее разделение можно проводить в термостатированном сатураторе Дэвиса со встроенным фильтром. Если равновесие проводится в отдельных сосудах, то фазы могут быть разделены с помощью пипетки с пористым фильтром или термостатированного ультрафильтра Тиссена под давлением от 2 до 3 атм. Хотя и трудно проводить центрифугирование точно при температуре равновесия, если нет термостатирующего помещения, все же кристаллизацию из горячих растворов можно предотвратить продуванием горячего воздуха через центрифугу и с помощью подогретых пипеток для удаления водной фазы. Фазы могут иногда быть отделены более полно, если прокладка из ваты центрифугируется на поверхности твердого вещества.
Выбор аналитического метода в основном зависит от величины измеряемой растворимости. Умеренно высокие значения обычно определяют гравиметрическим или объемным методом, а низкие значения - полярографическим, колориметрическим или радиометрическим методами. Незаряженные формы могут быть иногда отделены от насыщенного раствора экстракцией. Так, растворимость углеводородных лигандов в водных растворах серебра (I) была определена спектрофотометрически после экстракции лиганда гексаном, в то время как растворимость дитизона в буферных растворах измерялась добавлением избытка радиоактивного серебра, экстрагированием дитизоната серебра хлороформом и определением активности в органической фазе.
Хотя измерение растворимости обычно включает анализ насыщенного раствора, были выполнены также эксперименты по смещению ряда растворов известной концентрации с последующим определением веса образовавшегося осадка.
2. Прямой метод растворимости
В простейшем случае измерения растворимости применяются для изучения равновесия, когда единственными формами, присутствующими в обеих фазах (кроме растворителя и ионной среды), являются формы, образованные из центральной группы В и лиганда А.
Серьезным недостатком прямого метода растворимости является потеря одной степени свободы в насыщенных растворах, кроме того, метод ограничен системами комплексов, которые являются моноядерными относительно группы, произведенной целиком от труднорастворимого вещества.
Прямой метод растворимости особенно ценен для изучения кислот слишком нерастворимых (например, дитизон), чтобы их можно было исследовать другими методами. Однако для более растворимых веществ он менее удобен и, возможно, менее точен, чем потенциометрня. Этот метод имеет, кроме того, еще один недостаток: он не дает сведений об образовании полиядерных форм.
3. Метод конкурирующей растворимости
Если неудобно измерять растворимость ВАс, метод растворимости тем не менее может быть использован для определения констант устойчивости комплексов ВАn, при условии, что можно приготовить труднорастворимый твердый комплекс ВАС или ВАС, который содержит вспомогательную центральную группу В или лиганд А, и что можно определить независимо его произведение растворимости и константы устойчивости вспомогательного ряда комплексов ВАn. Метод конкурирующей растворимости в отличие от прямого метода в принципе может быть использован для изучения полиядерных комплексов ВqАp при условии, что начальные концентрации А и В могут меняться.
Ионный обмен
Катиониты являются полифункциональными соединениями, состоящими из высокомолекулярных анионов н простых катионов; промышленные синтетические вещества обычно являются формальдегидными или полистирольными смолами, которые содержат фенольную, сульфо- или карбоксильную группы в кислой форме или в виде соответствующей натриевой соли. Смолы не растворяются в воде и в большинстве органических растворителей. Если их привести в равновесие с раствором, содержащим ионы металла или другие катионы (например, ВАnz+ ), они могут участвовать в реакции обмена типа
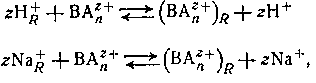
где подстрочная буква R обозначает фазу смолы. Нерастворимые амины или четвертичные аммониевые соли могут подвергаться подобным реакциям обмена с анионами в растворе, например,
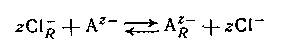
между анионным лигандом А и обменником в хлоридной форме. Большинство анионитов является несколько неустойчивыми смолами с высоким молекулярным весом, но на практике также применяются жидкие амины с умеренно низким молекулярным носом.
Из уравнений очевидно, что изучение полного распределения центральной группы В или лиганда А между ионообменииком и водной фазой может дать ценные сведения о формах, присутствующих в растворе. Еще в 1922 г. было проведено первое, хотя и безуспешное исследование комплексов металлов с помощью синтетического цеолита, но ионный обмен не применялся для изучения равновесия в растворе до конца 1945 г., когда стали легко доступными синтетические смолы. Как катиониты, так и аниониты использовались для определения природы форм, присутствующие в растворе, но обычно катиониты более пригодны для определения констант устойчивости.
1. Катионный обмен
Коэффициент распределения катиона ![]() где с+ - максимальное значение n для катионного комплекса) между водной фазой и натриевой формой катионита можно выразить через константу равновесия. Таким образом, стехиометрнческая константа распределения ВАnz+ определяется формулами
где с+ - максимальное значение n для катионного комплекса) между водной фазой и натриевой формой катионита можно выразить через константу равновесия. Таким образом, стехиометрнческая константа распределения ВАnz+ определяется формулами
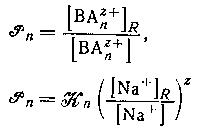 и будет постоянной при условии, что постоянны значения
и будет постоянной при условии, что постоянны значения ![]() и отношение концентрации ионов натрия в двух фазах. Последнее условие выполняется, если водная фаза содержит постоянную высокую концентрацию ионов натрия и обмен невелик. Подобным образом, если вероятен гидролиз группы В в смоле или в растворе, то достаточно использовать смолу в водородной форме и сильную кислоту в качестве фонового электролита. Использование постоянной ионной среды также обеспечивает постоянство коэффициентов активности в водной фазе.
и отношение концентрации ионов натрия в двух фазах. Последнее условие выполняется, если водная фаза содержит постоянную высокую концентрацию ионов натрия и обмен невелик. Подобным образом, если вероятен гидролиз группы В в смоле или в растворе, то достаточно использовать смолу в водородной форме и сильную кислоту в качестве фонового электролита. Использование постоянной ионной среды также обеспечивает постоянство коэффициентов активности в водной фазе.
Томпкинс и Мэйер нашли, что константа равновесия обмена между ионами лантана и аммония на смоле Дауэкс 50 достигает постоянной величины при очень низких концентрациях ионов лантана. Работы Фронеуса по ацетатным системам меди и никеля указывали на то, что при постоянной и очень малой загрузке смолы значения К зависят от концентрации свободных ацетат-ионов в водной фазе. Это означает, что, несмотря на разные заряды, формы В2+ и ВА+ действуют одинаково на коэффициенты активности в фазе смолы при условии, что они присутствуют только в небольших концентрациях. Поэтому Фронеус рекомендует получать количественные сведения о комилексообразованиии полной фазе на основе измерений, которые относились бы к постоянным и очень небольшим загрузкам смолы. Поэтому ионообменный метод не пригоден для количественного изучения систем, в которых образуются полиядерные формы. Для того чтобы обеспечить независимость констант ![]() от концентрации водородных ионов раствора, следует использовать сильнокислую однофункцнональную смолу, такую, как сульфированный полистирол (например, Дауэкс 50, Цеокарб 225 или Амберлит 120). Дальнейшим недостатком слабокислых смол, содержащих фенольные группы, является их тенденция к восстановлению поглощенных форм (например, иона VО2+ ). Если используются сильнокислые обменники при малой и постоянной загрузке, то коэффициенты активности в фазе смолы и отсюда стехиометрический коэффициент распределения между смолой и постоянной ионной средой будут оступаться постоянными.
от концентрации водородных ионов раствора, следует использовать сильнокислую однофункцнональную смолу, такую, как сульфированный полистирол (например, Дауэкс 50, Цеокарб 225 или Амберлит 120). Дальнейшим недостатком слабокислых смол, содержащих фенольные группы, является их тенденция к восстановлению поглощенных форм (например, иона VО2+ ). Если используются сильнокислые обменники при малой и постоянной загрузке, то коэффициенты активности в фазе смолы и отсюда стехиометрический коэффициент распределения между смолой и постоянной ионной средой будут оступаться постоянными.
Общее предположение, что только положительно заряженные формы сорбируются на катионите, было проверено на системе оксалата магния. Если оно справедливо в любом случае,- то распределение центральной группы между катионитом и раствором определяется выражением
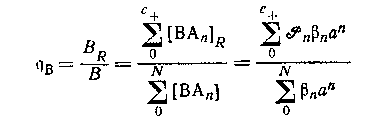
при условии, что коэффициенты активности в обоих фазах соответственно контролируются. Метод ограничивается системами комплексов катионных центральных групп с отрицательно заряженными лигандами.
Катионный обмен не является ни относительно точным, ни относительно удобным методом для определения констант устойчивости большинства систем. Функцию nв(а) невозможно определить с такой же точностью, которую часто получают в потенциометрии, а интерпретация данных включает (с++1) параметров в дополнение к искомым величинам βn.Так как В не может меняться в большой области концентраций, то этот метод ограничивается моноядерными системами. Более того, возникают заметные изменения nв от а, если лигандом является анион. Однако метод пригоден для изучения систем, в которых В следует сохранять очень низким (например, вследствие образования полиядерных форм при макроконцентрациях или из-за большой радиоактивности или недостаточного количества группы В). Наиболее удобно, когда происходит распределение только центральной группы, но для катионного обмена были получены обнадеживающие результаты, которые согласуются с данными других методов в системах с с+>0.
Анионный обмен
--> ЧИТАТЬ ПОЛНОСТЬЮ <--