Реферат: Влияние физических и химических факторов на основность алкиламинов
1.Кислоты и основания
Протон наиболее естественно воспринимается химиком как катион атома водорода"
Р.Белл
В современной химии используются две теории кислот и оснований: теория Бренстеда - Лоури и теория Льюиса - Усановича. Более общие определения кислот и оснований предложены в теории Льюиса - Уеановича. Однако в связи с важной ролью про-толитических процессов в химии теория Бренстеда - Лоури сохранила самостоятельное значение. Именно этой теории мы обязаны появлением комплекса проблем, ставших предметом настоящей книги.
1.1. Теория Бренстеда - Лоури
Согласно теории Бренстеда - Лоури кислота рассматривается как вещество, поставляющее протон, а основание - как вещество, способное присоединять протон (1,2)
![]() А В +Н + (1)
А В +Н + (1)
Кислота и отвечающее ей основание, таким образом, образуют сопряженную пару.
Ключевым в теории Бренстеда - Лоури является представление о том, что кислота взаимодействует при переносе протона с другой сопряженной парой (двойное протолитическое равновесие):
![]() А1 + В2 А2 + В1 (2)
А1 + В2 А2 + В1 (2)
Отсюда вытекают и первые предпосылки для количественного сравнения силы кислот и оснований: сильная кислота отдает протон ”легче”, чем слабая, а сильное основание “крепче” связывает протон, чем слабое. Равновесие тем полнее сдвинуто вправо, чем сильнее кислота А1 и слабее кислота А2 .
Можно представить, что двойное протолитическое равновесие является результатом двух сопряжённых равновесий:
![]() НА Н+ + А- (3)
НА Н+ + А- (3)
![]() В + Н+ ВН+ (4)
В + Н+ ВН+ (4)
Уравнение (4) лежит в основе количественной теории .позволяющей сравнивать силу различных оснований.
Безусловно, схема кислотно-основного равновесия, сформулированная Бренстедом и Лоури, не полностью отражает истинный процесс. Вернее, уравнение (4) следует считать адекватным процессам, происходящим в газовой фазе, что само по себе достаточно ценно и важно. В растворах же реакция между кислотой и основанием не сводится только к переносу протона от кислоты к основанию. Сначала кислота АН и основание В образуют комплекс АН...В за счет водородной связи между водородом кислоты и электронодонорным атомом основания. Во многих случаях протолитическая реакция преимущественно ограничивается этой начальной стадией комплексообразования. Поэтому такой процесс называется "незавершенным" кислотно-основным равновесием [4]. Следовательно, образование водородных связей рассматривается не только как вспомогательная, переходная ступень при кислотно-основном взаимодействии, облегчающая переход протона от кислоты к основанию, но и как один из самостоятельных видов этого взаимодействия. В благоприятных условиях кислотно-основное взаимодействие не останавливается на стадии комплекса АН...В, и происходит передача протона от кислоты к основанию, в результате чего основание протонируется. Эта вторая стадия цротолитического процесса называется "завершенным" кислотно-основным взаимодействием. При этом образовавшиеся ионы могут находиться в растворе либо в свободном виде, либо в виде ионных пар:
![]()
![]() ВН+ А- ВН+ | | А- ВН+ + А- , (5)
ВН+ А- ВН+ | | А- ВН+ + А- , (5)
где ВН+ А- - тесные ионные пары; ВН+ ççА- - сольватно-разделеные ионные пары ; ВН+ и А- -свободные ионы.
Более полным отражением кислотно-основного процесса является следующая схема [4]:
![]()
![]()
![]() АН + В а АН...В б А- ...ВН+ в А- + ВН+ , (6)
АН + В а АН...В б А- ...ВН+ в А- + ВН+ , (6)
здесь а - незавершенное кислотно-основное равновесие, б - завершенное и в - диссоциация на свободные ионы* . Таким образом, количественное сравнение слабых оснований в рамках теории Бренстеда - Лоури должно осуществляться с учетом реальной ситуации, в которой происходит перенос протона к органическому основанию: в газовой фазе основой для количественных расчетов основности может служить принципиальная схема (4), тогда как в растворах следует опираться на схему (6), а вернее, на ее модификации, учитывающие конкретные условия.
1.2. Физический смысл и меры основности в газовой фазе
Основностью в газовой фазе называют свободную энергию (DG) равновесия (4) [5,6] . Как известно, DG = DН0 - ТDS. Измерения энтропии равновесия в газовой фазе показали, что эта величина обычно не превышает 9-12 Дж/(моль·К). Таким образом, изменение энтальпии равновесия (DН0 ) считается равным (DG0 ) [5].
Изменение энтальпии равновесия (4) переноса протона в газовой фазе, взятое с обратным знаком, называется сродством к протону и обозначается РА (Proton Affinity). Численное значение РА определяется из соотношения:
РА=-DН0 =DН0 (Н+ )+ DН0 (В) - DН0 (ВН+ ) (7)
Где DН0 (Н+ ) - энтальпия образования иона Н+ ; DН0 (В) и DН0 (ВН+) - энтальпия образования основания (В) и его протонированной формы (ВН+ ) соответственно. DН0 (Н+ ) = 1530 кДж. Энтальпии образования органических соединений известны из термохимических справочников или могут быть вычислены по соответствующим эмпирическим формулам [7]. Следовательно, определение сродства к протону РА зависит от возможности определения энтальпии образования протонированной формы основания DН0 (ВН+ ).
Экспериментально определение сродства оснований к протону осуществляется с помощью спектроскопии ионного циклотронного резонанса [8] и масс-спектрометрии высокого давления [5. В настоящее время эти методы позволяют вычислять значения РА с точностью ±0,9 кДж/моль. Таким образом, величина РА является надежным критерием для сравнения основности соединений в газовой фазе.
Существует мнение [5], что на базе значений РА можно построить ”абсолютную”” шкалу основности органических соединений. В последние годы получены значения РА для некоторых алифатических и ароматических аминов, гетероциклических соединений, спиртов, эфиров, альдегидов, кетонов, карбоновых кислот [5] и нитросоединений [9].
2. Основность аминов в газовой фазе
Из рассмотрения данных по основности аминов в растворах следует, что она существенно зависит от электронных и пространственных факторов структуры и сольватации исследуемых соединений. Несмотря на отмеченные успехи при выявлении отдельных зависимостей между основностью и указанными эффектами, до сих нет единого количественного подхода к решению проблемы в целом. Казалось бы, что для оценки влияния строения аминов на их основность наиболее удобными есть соответствующие характеристикистики в газовой фазе, в которой, естественно, сольватационные эффекты отсутствуют. С этой точки зрения большой интерес представляет серия появившихся в последнее время работ (в том числе и обзорных [3, 6, 7, 21,137—140]), в которых обобщены результаты масс-спектрометрических методов, в которых определялись энергии перехода протона между стандартным и рассматриваемым (В) аминами в газовой фазе:
![]() В0 Н+ + В В0 + ВН + (1)
В0 Н+ + В В0 + ВН + (1)
Наличие свободной энергии этого процесса характеризует основность амина В относительно стандарта В0 . Абсолютная основность (В0 ), равная отрицательной свободной энергии (DG0 ) процесса (8), может быть рассчитана, если известно соответствующее
В + Н+ =ВН+ (2)
абсолютное значение для стандартного основания. Энтальпия (DH0 ) процесса (8), взятая с обратным знаком, характеризует сродство аминов к протону (РА) и легко рассчитывается, так как для прото-нирования аммиака, обычно принимаемого за стандарт, она определена независимыми методами [139, 143, 148, 149, 150]. Абсолютные значения 0В и РА* зависят от выбранного стандарта и абсолютной величины РА для него (например, для аммиака значение РА изменяется достаточно широко: 200,7 [139]; 201,4 [151]; 202,3 ± 2,0 [152]; 207 ± 3 [143, 148, 150, 153], 211,3 [149]; 214,4 [45]). Поэтому для выяснения количественных закономерностей влияния структуры аминов на их основность в газовой фазе лучше всего пользоваться величинами DGB (или — dR DG0 [3,7, 47]), когда за стандарт выбран аммиак**.
В табл.1 приведены величины DGB известного к началу 1977 г. ряда аминосоединений. Сопоставление этих данных показывает, что поведение различных аминов как оснований в газовой фазе резко отличается от такового в конденсированных средах. Например, анилин (табл. 1, № 31) в газовой фазе оказался на 6,7 ккал/моль (или почти на 5 ед. рКа ) более основным, чем аммиак (№ 1), в то время как в воде, нитрометане и ацетонитриле (см. выше) наблюдается противоположная ситуация. Аналогичная картина имеет место и для пиридина, который в конденсированной фазе примерно на 4 ед. рКа менее основен, а в газовой фазе (№ 80) на 16 ккал/моль (или ~ на 12 ед. рКа ) более основен, чем аммиак; для ацетамида (№ 32), который в воде ~ на 10 ед. рКа менее основен по сравнению с аммиаком, а в газовой фазе практически равен ему; для пиррола (№ 54) и некоторых других аминосоединений.
Таблица 1. Значения основностиа аминосоеденений в газовой фазеб относительно аммиака
| Номер | Амин | DGBг) | ||
| 1 | NH3 | 0,0 | ||
| 2 | CH3 NH2 | 9,1 | ||
| 3 | C2 H5 NH2 | 11,8 | ||
| 4 | n-C3 H7 NH2 | 13,0 | ||
| 5 | i-C3 H7 NH2 | 14,1 | ||
| 6 | n-C4 H9 NH2 | 13,5 | ||
| 7 | i-C4 H9 NH2 | 14,0 | ||
| 8 | s-C4 H9 NH2 | 15,2 | ||
| 9 | t-C4 H9 NH2 | 16,1 | ||
| 10 | n-C5 H11 NH2 | 13,4д ) | ||
| 11 | t-C5 H11 NH2 | 17,4 | ||
| 12 | n-C6 H13 NH2 | 13,5д ) | ||
| 13 | n-C7 H15 NH2 | 13,6д ) | ||
| 14 | c-C6 H11 NH2 | 16,3 | ||
| 15 | NH2 -NH2 | 3,8 | ||
| 16 | NH2 (CH2 )2 NH2 | 19,0д ) | ||
| 17 | NH2 (CH2 )3 NH2 | 24,5д ) | ||
| 18 | NH2 (CH2 )4 NH2 | 27,1д ) | ||
| 19 | NH2 (CH2 )5 NH2 | 25,4д ) | ||
| 20 | NH2 (CH2 )6 NH2 | 25,4д ) | ||
| 21 | NH2 (CH2 )7 NH2 | 25,2е ) | ||
| 22 | CH3 O(CH2 )2 NH2 | 14,7д ) | ||
| 23 | H2 C=CH-CH2 NH2 | 11,3 | ||
| HCºC-CH2 NH2 | 6,7 | ||
| 25 | NCCH2 CH2 NH2 | 3,0 | ||
| 26 | CF3 (CH2 )3 NH2 | 10,1 | ||
| 27 | FCH2 CH2 NH2 | 8,0 | ||
| 28 | CF3 (CH2 )2 NH2 | 6,7 | ||
| 29 | F2 CHCH2 NH2 | 4,0 | ||
| 30 | CF3 CH2 NH2 | -1,4 | ||
| 31 | 6,8 | |||
| 32 | CH3 CONH2 | 0,2ж ) | ||
| 33 | HCONH2 | -7,1з ) | ||
| 34 | (CH3 )2 NH | 15,5 | ||
| 35 | CH3 NHC2 H5 | 17,9 | ||
| 36 | (C2 H5 )2 NH | 20,2 | ||
| 37 | (n-C3 H7 )2 NH | 22,2 | ||
| 38 | (i-C3 H7 )2 NH | 23,9 | ||
| 39 | (n-C4 H9 )2 NH | 23,1 | ||
| 40 | (i-C4 H9 )2 NH | 23,6 | ||
| 41 | (s-C4 H9 )2 NH | 25,8 | ||
| 42 | 11,2 | |||
| 43 | 18,0 | |||
| 44 | 20,1 | |||
| 45 | 21,2 | |||
| 46 | 19,2д ) | |||
| 47 | 14,4д ) | |||
| 48 | (H2 C=CHCH2 )2 NH | 19,3 | ||
| 49 | (HCºCCH2 )2 NH | 11,7 | ||
| 50 | NCCH2 NHCH3 | 2,7з ) | ||
| 51 | CF3 CH2 NHCH3 | 6,2з) | ||
| 52 | 12,9 | |||
| 53 | 15,3ж) | |||
| 54 | 4,0ж) | |||
| 55 | NHMe-C=O H | 1,7 | ||
| 56 | (CH3 )3 N | 20,0 | ||
| 57 | (CH3 )2 NC2 H5 | 22,4 | ||
| 58 | (C2 H5 )2 NCH3 | 24,6 | ||
| 59 | (C2 H5 )3 N | 26,7 | ||
| 60 | (C3 H7 )3 N | 28,7 | ||
| 61 | 17,1и) | |||
| 62 | 8,1и) | |||
| 63 | 24,3 | |||
| 64 | 25,7 | |||
| 65 | 27,1 | |||
| 66 | 26,1к) | |||
| 67 | (CH3 )2 N-NH2 | 15,2 | ||
| 68 | ((CH3 )2 NCH2 )2 | 30,3 | ||
| 69 | 23,5 | |||
| ||||
70 | (H2 C=CH-CH2 )3 N | 24,7 | ||
| 71 | (HCºC-CH2 )3 N | 15,0 | ||
| 72 | NCCH2 N(CH3 )2 | 7,1 | ||
| 73 | F3 CCH2 N(CH3 )2 | 20,9 | ||
| 74 | 19,5 | |||
| 75 | 21,0ж ) | |||
| 76 | 23,7ж ) | |||
| 77 | 19,3 | |||
| 78 | 21,8 | |||
| 79 | 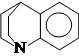 | 26,0 | ||
| 80 | 16,0 | |||
| 81 | CH3 CON(CH3 )2 | 11,7 | ||
| 82 | HCON(CH3 )2 | 7,6 | ||
| 83 | NF3 | -56л) |
Существенные различия между свойствами в газовой и конденсированной фазах наблюдается и при сравнении оснований одного и того же класса. Например, все первичные алкиламины в газовой фазе (№2—29), за исключением b,b,b-трифторэтиламина (№30), оказались более основными, в то время как в воде (см. например, табл. 1) амины с электроотрицательными заместителями зачастую менее основны, чем аммиак. То самое относится и ко вторичным и третичным алкиламинам.
Данные по изменению свободной энергии и энтальпии реакций, описываемых уравнениями (17) — (2), совместно с некоторыми другими результатами позволили определить термодинамические характеристики процессов переноса свободных и протонированпых оснований из газовой фазы в водные растворы и на этой основе про вести термодинамический анализ влияния сольватации на основность аминов в воде [3, 6, 47, 140, 151, 153]. При этом преимущественное внимание было уделено причинам, обусловливающим наблюдаемый порядок изменения основностии в воде при переходе от аммиака к первичным, вторичным и третичным алкиламинам с насыщенными углеводородными заместителями. На основе этих данных Ариетт с сотрудниками [3, 6, 47] сделал вывод, что главным фактором, определяющим наблюдаемый порядок основности аминов различных классов в воде, является специфическая сольватация соответствующих катионов, зависящая от числа атомов водорода у протонированного азота. Неспецифическая же сольватация, по их мнению, имеет второстепенное значение, т. е. эти исследователи придерживаются сольватационной (гидратационной) теории Тротмана — Диккенсона (см. выше).
В то же время другая группа исследователей [140, 153] считает, что изменение основности аминов при переходе из газовой фазы в воду обусловлено прежде всего электростатической (неспецифической) сольватацией катионов, а специфическое взаимодействие играет второстепенную роль. При этом в указанных работах принимается, что кислотно-основные свойства соединений в газовой фазе являются истинными (собственными) свойствами, и в противоположность случаю в конденсированной фазе практически не обсуждается зависимость этих свойств от строения аминов.
--> ЧИТАТЬ ПОЛНОСТЬЮ <--