Реферат: ЯМР как аналитический метод
Введение
В этой работе на разнообразных примерах показано, каким образом ЯМР может быть использован в качестве аналитического метода. С этой целью более подробно обсудим некоторые понятия.
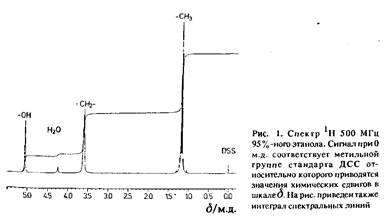
Применение метода ЯМР в качестве аналитического можно продемонстрировать на примере простейшей молекулы – молекулы этанола, поскольку, как видно из рис. 1, протонам трех функциональных групп в этаноле: метильной, метиленовой и гидроксильной соответствуют три различные резонансные линии, наблюдаемые в спектре. Особенность метода ЯМР прежде всего состоит в том, что по положению резонансных линий в спектрах можно судить о взаимном расположении отдельных атомов или групп атомов в молекулах, причем это удается обнаружить даже для эквивалентных атомов. ЯМР по своей информативности выгодно отличается от многих других аналитических методов, конкурирующих с ним.
В то же время ЯМР является довольно малочувствительным методом, несмотря на то, что в современных спектрометрах значение максимально достижимого магнитного поля составляет 14 Тл, что соответствует рабочей частоте для 1 H 600 МГц. Минимальное количество вещества, которое может быть зарегистрировано с помощью метода ЯМР 1 H, варьируется в широких пределах в зависимости от условий эксперимента. Для малых молекул типичное значение, определяющее предел чувствительности, составляет 10 нмоль, т.е. то количество вещества, которое может быть проанализировано простыми биохимическими методами, например, с помощью тонкослойной хроматографии. При использовании других, достаточно широко используемых методов, таких, как газовая хроматография или радиоиммунометрия, становятся доступными принципиально иные с точки зрения чувствительности области такие, как фемто- и атоммолярная. Таким образом, если речь идет только о регистрации спектров веществ известной структуры, то эти методы по чувствительности превосходят ЯМР. Преимущество ЯМР становится очевидным лишь тогда, когда возникает необходимость в получении дополнительной информации, которую может предоставить только этот метод. Это касается тех случаев, когда структура исследуемого вещества еще не известна. Тогда из анализа спектра ЯМР можно построить ряд возможных структур и выбрать среди них правильную. Следует отметить также еще одну особенность метода ЯМР: в спектрах ЯМР 1 H могут присутствовать сигналы молекул-примесей, идентификация которых возможна только в случае применения специальных методов.
1. Идентификация известных и неизвестных веществ
Использование метода ЯМР в качестве аналитического в простейшем случае состоит в сравнении положений линий спектров образца неизвестной структуры и стандартного образца известной структуры: совпадение спектров стандартного вещества и исследуемого позволяет провести идентификацию последнего. На основании такого рода сопоставлений можно дать даже количественную оценку концентрации веществ, содержащихся в образце. Эта оценка в простейшем виде может быть сформулирована следующим образом: вещество А содержится в образце в концентрации, достаточной для его регистрации. Правда, такой способ оценки справедлив лишь тогда, когда оба сравниваемых спектра получены при идентичных условиях. Реальный спектр, соответствующий определенному веществу, существенно зависит от условий его регистрации. Вид спектра определяется не только внешними факторами, т.е. теми параметрами, которые экспериментатор может контролировать самостоятельно, но и используемым растворителем. Несмотря на это, все же удается провести идентификацию неизвестного вещества, поскольку всегда можно ограничиться определенным абстрактным уровнем и сравнить характерные особенности спектров.
Рассмотрим еще раз фрагмент спектра ЯМР этанола, снятого на современном ЯМР-спектрометре с высоким значением постоянного магнитного поля. Параметры, полностью характеризующие спектр, следующие: положение спектральных линий, структура мультиплетов и относительные интенсивности резонансных линий. Положение линий определяется значением химического сдвига, а расщепление линий – скалярным спин-спиновым взаимодействием. Площадь под резонансным сигналом, которая может быть получена численным интегрированием, пропорциональна числу спинов, соответствующих данной группе в молекуле.
Как было показано ранее, химический сдвиг можно понимать как усиление или ослабление внешнего магнитного поля в точке пространства, в которой расположено исследуемое ядро. Основной вклад в величину этого локального изменения поля дают электронные оболочки рассматриваемых молекул, поэтому можно утверждать, что химический сдвиг отражает структуру молекул. Если оценивать влияние заместителей на величину химического сдвига, то с ростом числа связей между рассматриваемым атомом и заместителем это влияние уменьшается. Однако характер электронного распределения, а значит, и вид спектра ЯМР, зависит и от пространственной структуры молекул, и от взаимодействия с другими молекулами в растворе, т.е. от тех факторов, которые уже не представляются столь очевидными.
Для ЯМР область значений химических сдвигов невелика, а эффекты, связанные с пространственной структурой и вызванные взаимодействием с другими молекулами, бывают настолько велики, что часто из наблюдения спектров ЯМР весьма затруднительно сделать однозначный вывод о химической структуре вещества.
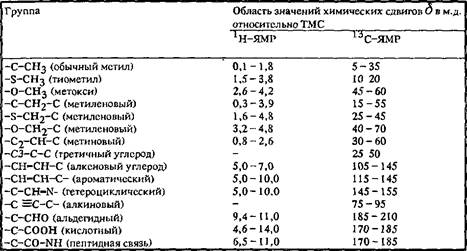
Таблица 1. Химический сдвиг в спектрах Ни С
Для большинства молекул в растворе конформация их при комнатной температуре не является жесткой, они могут существовать в виде нескольких различных конформаций. Устанавливаемое конформационное равновесие зависит от состава растворителя, температуры и давления. Кроме того, исследуемые молекулы могут взаимодействовать между собой или с другими молекулами и образовывать при этом более или менее стабильные комплексы. Вероятность обнаружения этих взаимодействий зависит от концентрации всех компонентов, присутствующих в растворе.
В отличие от спектров ЯМР 1 H диапазон значений химических сдвигов в спектрах С существенно больше», так что положение резонансных линий в спектрах 13 C определяется в основном химической структурой. Значения химических сдвигов характеристичны при условии, что влияние заместителей учитывается в виде аддитивной поправки к величине химического сдвига.
2. Внутренний и внешний стандарт
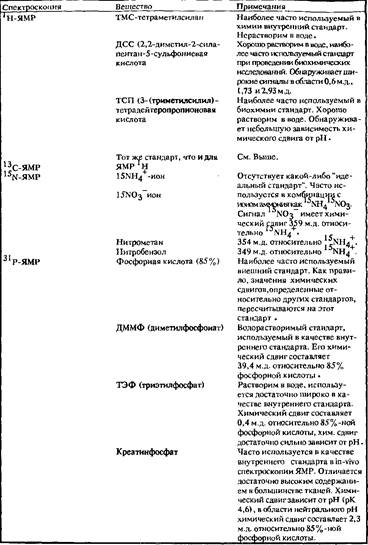
Химический сдвиг, как правило, приводится относительно резонансной частоты вещества-стандарта, которое добавляется в исследуемый раствор или помещается в капилляр. В этом случае, как показано ранее, химический сдвиг зависит от резонансной частоты используемого стандарта. Величина химического сдвига имеет смысл только в случае, если указан стандарт, относительно которого приводится его значение. В табл. приведены основные вещества, используемые в качестве стандартов. Если же химический сдвиг приведен относительно другого вещества, используемого в качестве стандарта, то его значение всегда можно пересчитать относительно общепринятого стандарта.
Как внешний, так и внутренний стандарты обладают рядом преимуществ и недостатков. Внутренний стандарт может вступать во взаимодействие с другими молекулами, присутствующими в растворе, что приводит к сдвигу сигнала стандарта, т.е. к сдвигу нулевой точки. Взаимодействие стандарта с исследуемым веществом также может привести к изменению вида его спектра. Поэтому основным требованием, предъявляемым к внутреннему стандарту, является уменьшение этого эффекта. Очевидно, что внутренний стандарт должен быть химически стабильным соединением и хорошо растворимым в используемом растворителе. Всем этим условиям вполне удовлетворяют такие вещества, как тетраметилсилан, 2,2 – диметил‑2‑силапентан‑5‑сульфоновая кислота и триметилсилилпропионовая кислота, положение сигнала метальной группы для которых используется в качестве нулевой точки в шкале химических сдвигов и м.д. Эти вещества используются в качестве стандартов в спектрах как 1 H, так и 13 C Для других ядер, спектры которых представляют интерес с точки зрения биологических приложений ЯМР таких, как 15 N и 31 P, пока не установлен единый стандарт.
Внешний стандарт отделен от исследуемого вещества. Как правило, его запаивают в капилляр и помещают в измерительную ампулу. Очевидным преимуществом использования внешнего стандарта является то, что молекулы стандартного вещества и исследуемого не взаимодействуют. Однако здесь возникает другая проблема, связанная с тем, что магнитные восприимчивости растворителя внутри капилляра и в образе различаются. Величина магнитного поля в точке расположения ядра определяется не только экранирующим влиянием электронных оболочек, но и внешним магнитным полем B0 , величина которого зависит от магнитной восприимчивости окружения. Если используется внутренний стандарт, то изменение магнитного поля за счет восприимчивости окружения одинаково и для молекул стандарта, и для молекул исследуемого вещества. При использовании внешнего стандарта это условие не выполняется. В зависимости от магнитных восприимчивостей исследуемого вещества и стандарта их ядра находятся в различных магнитных полях, причем это различие в величине полей зависит не только от магнитных восприимчивостей![]() исследуемого вещества и
исследуемого вещества и![]() вещества-стандарта, но и геометрических факторов. В ЯМР высокого разрешения образец и стандарт
вещества-стандарта, но и геометрических факторов. В ЯМР высокого разрешения образец и стандарт
помещают, как правило, в цилиндрические ампулы-капилляры, длинные оси которых параллельны внешнему магнитному полю. Различие в значениях внешнего магнитного поля для образца и стандарта можно рассчитать по следующей формуле:
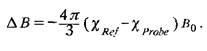
Поправка на восприимчивость оказывается особенно существенной, если проводится сравнение спектров, полученных на спектрометре со сверхпроводящим магнитом и с обычным электромагнитом. В последнем случае образцы обычно располагают перпендикулярно внешнему магнитному полю; согласно теоретическим расчетам коэффициент –4/3 в уравнении следует заменить на 2/3. Таким образом, даже для одинаковых образцов без учета поправки на магнитную восприимчивость значения химических сдвигов будут отличаться.
Для образца сферической формы магнитное поле внутри и вне его одинаково, так что проблему, связанную с внешним стандартом, можно частично обойти, если поместить стандарт в сферический капилляр.
3. Мультиплетная структура
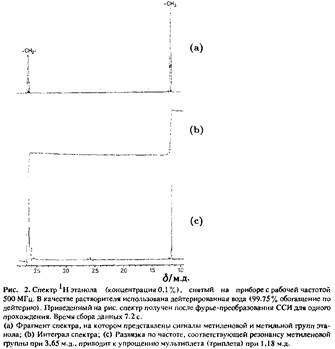
Вторым важным свойством спектров ЯМР, позволяющим получить более полную их характеристику, является мультиплетная структура линий. Параметр J, количественно характеризующий расщепление резонансных линий, называется константой связи. Величина / называется константой гомоядерного взаимодействия, если рассматривается взаимодействие между ядрами одного сорта, и константой гетероядерного взаимодействия, если взаимодействующие ядра принадлежат ядрам различных элементов. Расщепление резонансных линий, показанное на рис. 1, отражает гомоядерное взаимодействие между протонами, которые входят в состав различных химических групп в этаноле. Этанол состоит не только из атомов водорода, но и атомов углерода и кислорода, однако константа гетероядерного взаимодействия не может быть обнаружена по спектрам ЯМР, поскольку изотопы 12 C и 16 O обладают нулевым спином. При более детальном рассмотрении спектра можно наблюдать очень слабые дополнительные линии по обе стороны от основной – так называемые сателлиты С. Эти линии возникают за счет взаимодействия протонов с изотопом углерода С, естественное содержание которого составляет порядка 1% и спин 7=1/2.
Мультиплетная структура спектров ЯМР может быть рассчитана теоретически. Разработан ряд достаточно совершенных программ построения спектров, которые позволяют по известным значениям констант спин-спинового взаимодействия и химических сдвигов провести расчет теоретического спектра. В случае слабой связи характер расщепления можно описать достаточно просто, и наоборот, исходя из характера расщепления резонансных линий, можно рассчитать, сколько спинов участвует во взаимодействии и какова структура данного химического соединения. При смешивании этанола с дейтерированной водой протоны гидроксильной группы этанола замещаются на ядра дейтерия. В спектре исчезает сигнал, соответствующий протонам гидроксильной группы и одновременно исчезает взаимодействие протонов гидроксильной группы и протонов CH2 -группы. Протоны, входящие в состав CH2 - и СН3 -групп в пределах группы магнитно эквивалентны. Это соответствует спиновой системе A2 X3 , спектр которой приведен на рис. 2: расщепление резонансных линий CH2 -группы – квартет, а СН3 -группы – триплет.
Интенсивность компонент мультиплета также можно рассчитать по простым правилам, аналогичным правилам расчета мультиплетной структуры. В принципе с помощью итеративной процедуры можно получить соотношение интенсивностей линий внутри мультиплета для каждой резонансной линии. Взаимодействие данного спина с п эквивалентными ядрами со спином 1/2 вызывает расщепление резонансной линии на компоненту, интенсивность каждой из компонент описывается
биномиальными коэффициентами ![]() . Интенсивность Im т-н компоненты в этом случае можно рассчитать по формуле
. Интенсивность Im т-н компоненты в этом случае можно рассчитать по формуле
--> ЧИТАТЬ ПОЛНОСТЬЮ <--