Реферат: Ионные реакции в растворах. Солевой эффект (в ТАК)
![]()
![]()
![]() (6.7)
(6.7)
2.3) Это же получается для бимолекулярной реакции и при стандартизации концентрации:
 (6.8)
(6.8)
![]()


![]()

в бимолекулярном акте активации n# = -1, и![]() (6.10)
(6.10)
Результат: Формула, связывающая энергию активации Аррениуса с квазитермодинамическими функциями активации теории переходного состояния, не зависит от выбора стандартного состояния.
3. Адиабатические потенциалы и потенциальные поверхности
Пример. Реакция обмена одного из атомов в молекуле водорода на дейтерий
(Это простейший из любых возможных примеров)
![]()
По мере сближения атома дейтерия с молекулой водорода наблюдается разрыхление старой двухцентровой химической связи H-H и постепенное оформление новой связи H-D, так что энергетическая модель реакции дейтерообмена в молекуле водорода может быть построена как постепенное перемещение исходной трёхатомной системы к конечной согласно схеме: 
К ним могут относиться превращения разной природы: и окисление-восстановление, и обмен лигандами между комплексными ионами, и их объединяет кинетическая схема вида:
![]() .
.
Первая стадия равновесная, а за нею следует медленное превращение в продукты. Константа скорости в ТАК, равна ![]() . (10.1)
. (10.1)
Она была построена для смеси идеальных газов и согласно принципу детального равновесия содержит константу равновесия Kc# первой стадии образования активированного комплекса. Через неё в активационное соотношение вводятся концентрации реагентов, но квазитермодинамические активационные функции H# и S# для неидеальной смеси реагентов полагается связать уже не с Kc#, а с Ka#. При переходе к неидеальным системам необходимо скорректировать полученное в ТАК выражение константы скорости:
 (10.2)
(10.2)

Искомая коррекция выражается уравнением Брёнстеда-Бьеррума в виде:
 или в логарифмической форме
или в логарифмической форме 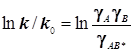 . (10.3)
. (10.3)
С его помощью были исследованы некоторые реакции обмена лигандами между комплексными ионами переходных металлов (d-элементов). Скорости этих реакции обычно сравнительно невелики и доступны для традиционных классических методов измерений. Обратимся к теории растворов сильных электролитов по Дебаю- Хюккелю.
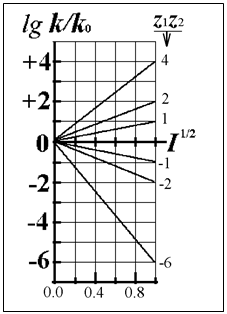
Для миллимолярных (почти «предельно разбавленных») растворов справедливо:
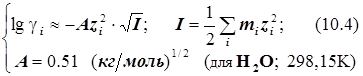
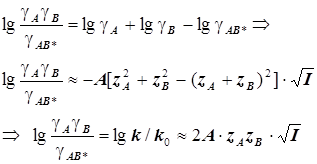
![]()
Константы скоростей согласно формуле (8.5) линейно зависят от квадратного корня из ионной силы раствора. Графики этих зависимостей (точнее, касательные к ним) образуют пучок прямых, и значения их угловых коэффициентов оказываются дискретными - «квантованными», поскольку заряды ионов zi и их произведения zAzB принадлежат к ряду целых (и положительных, и отрицательных) чисел. (рис.21.) Это кинетическое явление называется первичным солевым эффектом.

Реакции в растворах (дополнения)
Особенно интересны реакции, которые можно изучить и в газовой фазе, и в растворе. Их немного: