Реферат: Равновесия в неводных растворах
где VM — мольный объем; а М — истинная молекулярная масса.
2. Образование продуктов присоединения (сольватация)
Переходя к рассмотрению следующей стадии равновесий в растворах, связанной со взаимодействием между компонентами раствора, необходимо, хотя бы кратко, остановиться на определении понятия «химическое взаимодействие в растворе».
Ответ на вопрос, что следует считать необходимым и достаточным признаком для суждения об отсутствии химического взаимодействия между компонентами раствора с самого начала был связан с трудностями методологического характера. Как известно, не может быть дано сколь-нибудь строгого определения негативного понятия, а отсутствие взаимодействия несомненно принадлежит к числу таковых.
Далее, если считать признаком возникновения химической связи взаимодействие электронных облаков атомов химических элементов, то строго говоря, поскольку плотность электронного облака на любом конечном расстоянии от атомного ядра не равна нулю, следует признать наличие химического взаимодействия между любыми двумя атомами, находящимися друг от друга на любом конечном расстоянии. Но, очевидно также и то, что исповедование этого положения лишило бы химию сколь-нибудь четких критериев в определении самого ее фундаментального понятия. Поэтому в определении понятия химическое взаимодействие (так же, как и атомный радиус) всегда лежит какое-либо предварительно оговоренное условие, а само оно всецело зависит от этого условия.
По-видимому, наиболее удобным критерием, который мог бы служить для суждения о наличии или отсутствии химического взаимодействия в химической системе, является энергия взаимодействия. Можно было бы выбрать такую минимальную ее величину, которая могла бы быть отнесена к химическому процессу. Однако несовершенство и значительная сложность методов определения энергии взаимодействуя не позволяет в настоящее время считать такой выбор удачным.
Тип связи также не может служить критерием, который лег бы в основу суждения о наличии или отсутствии взаимодействия. В самом деле, между крайними случаями — образованием чисто ковалентной или ионной связи и слабым вандерваальсовским взаимодействием — существует множество промежуточных типов, одна классификация которых подчас вызывает значительные затруднения.
Строго говоря, процесс молекулярной ассоциации (диссоциации) должен быть отнесен к химическому взаимодействию. Действительно, если, например, ассоциат (АН)Х через Н-связь подвергается молекулярной диссоциации, то это сопряжено с разрывом Н-связи, т. е. по всем формальным признакам (в число которых включается и весьма значительная величина энергии связи) подходит под химическое взаимодействие. Однако ассоциативно-диссоциативные процессы не принято относить к химическому взаимодействию. В противном случае пришлось бы разграничивать химическое взаимодействие, относящееся к первой стадии равновесий в растворах и межмолекулярное взаимодействие между компонентами раствора:
![]()
(предполагается для простоты, что молекулы растворителя также ассоциированы через Н-связь).
.Впрочем, вряд ли было бы целесообразным считать любое межмолекулярное взаимодействие данного типа химическим. Так, если АН и SH — соединения тождественной химической природы, стоящие к тому же близко в гомологическом ряду (например, пропанол и бутанол, изомеры крезола, валериановая и масляная кислоты и т. п.), то образование новых межмолекулярных связей (перекрестных ассоциатов) не может быть отнесено к химическому взаимодействию, хотя бы потому, что это взаимодействие практически не сопряжено с таким изменением энергетических характеристик системы, которое могло бы быть с уверенностью зафиксировано.
Выбор критерия, которым следует пользоваться для суждения о взаимодействии в химической системе, в каждый данный период развития химии зависит от развития теория, а также методов исследования химических систем. Так, в настоящее время благодаря интенсивному развитию ИК- и радиоспектроскопии, появляется возможность получения гораздо более интимных сведений о химической системе, чем два-три десятилетия назад. И несомненно, что спустя определенный промежуток времени появятся методы исследования, которые углубят степень познания процессов, протекающих в системе, столь же значительно, как это сделали упомянутые методы. Вот почему всегда, говоря о наличии или отсутствии химического взаимодействия, следует оговаривать, о какой степени приближения идет речь, так как очевидно, что универсального определения, одинаково пригодного для всех случаев, быть не может.
Хотя современные методы исследования позволяют зафиксировать весьма слабые химические взаимодействия с энергией порядка десятой доли ккал/моль, в подавляющем большинстве случаев в растворах они не могут быть с уверенностью обнаружены. Дело в том, что энергетические эффекты смешения соизмеримы, а зачастую значительно превышают указанную величину. Вот почему слабая энергетика химического взаимодействия в растворах не может быть выделена на фоне значительных тепловых эффектов смешения, а также теплового движения молекул.
В этих случаях предпочитают говорить'об отсутствии химического взаимодействия между компонентами, образующими раствор. Такая точка зрения является приближением, весьма пригодным для рассмотрения процессов, протекающих в растворах.
Таким образом, процесс химического взаимодействия между растворенным веществом АК и растворителем S
![]()
с точки зрения -представлений, которые будут соблюдаться и в этой книге, протекает не всегда. Он необходим лишь тогда, когда образуется электролитный раствор. В литературе химическое соединение (АК)m Sn часто называют сольватом, а сам процесс — сольватацией. Прежде понятие «сольват» очень часто отождествляли с понятием «соединение неопределенного состава», В тех же случаях, когда стехиометрия взаимодействия между АК и S была точно известна, употребляли термин «продукт присоединения». По мере усовершенствования методов исследования растворов смысловое различие между этими двумя понятиями постепенно исчезает. В этой книге мы будем считать эти понятия синонимами, применяя, следуя традиции, укоренившейся в литературе, термин «сольватация» для обозначения процессов присоединения молекул растворителя к ионам, а термин «продукт присоединения» — для сольватов, образующихся в жидких системах. В случае ионных сольватов принято различать первичную и вторичную сольватные оболочки. При этом в первичную оболочку входит то сравнительно небольшое количество молекул растворителя, которые непосредственно связаны с ионом (причем энергия связи ион — молекула растворителя соизмерима с энергией химической связи) и которые перемещаются в растворе вместе с ионом. Вторичная сольватная оболочка включает значительно большее число молекул растворителя. Границы этой оболочки определяются изменением физико-химических характеристик растворителя под влиянием иона.
Исследование стехиометрии и глубины взаимодействия в растворах, т. е. изучение стехиометрии и константы равновесия процесса является предметом обширнейшего раздела теории растворов.
3. Ионизация
Энергетический анализ процесса электролитической диссоциации показывает, что сольват (AK)m Sn не может непосредственно перейти в диссоциированные на ионы соединения. Так, продукт присоединения амина к кислоте RCOOH • NR3 ’ не может непосредственно дать ионы RCOO- и [NR3 H]+ . Для этого потребовалось бы затратить весьма значительную энергию, необходимую для разрыва связи —О-Н. Поэтому стадии электролитической диссоциации продукта присоединения амина к кислоте предшествует, стадия ионизации продукта присоединения, заключающаяся в отторжении протона от гидроксильной группы с переходом его на атом азота, причем образуется ионизированный комплекс RCOO- -[NR3 H]+ .
В общем виде процесс ионизации передается схемой:
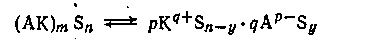
Нередко взаимодействие между компонентами раствора может ограничиваться стадией ионизации. Особенно часто это встречается в растворах с низкой диэлектрической проницаемостью. Например, при сливании разбавленных бензоловых растворов трихлоруксусной кислоты и триэтиламина образуется практически нацело ионизованный продукт присоединения, однако электропроводность раствора очень низка.
Наиболее удобным методом изучения стадии ионизации в растворах является исследование колебательных спектров. Однако константа ионизации при этом может быть рассчитана лишь тогда, когда электролитическая диссоциация протекает в малой степени, так как в подавляющем большинстве случаев на ИК-спектрах полосы поглощения, отвечающие ионам, находящимся и в свободном состоянии и в ионизованном комплексе, неразличимы.
В жидких системах информация о стадии ионизации может быть получена на основании кондуктометрических измерений.
В растворителях с высокими диэлектрическими проницаемостями электролитическая диссоциация может протекать полностью и равновесная концентрация ионизованного комплекса будет пренебрежимо мала. Однако это не означает, что электролитный раствор мог образоваться, минуя стадию ионизации. Развитие методов изучения быстрых реакций показывает, что даже в растворителях с весьма высокими диэлектрическими проницаемостями (вода, серная кислота) возникновению свободных ионов неизбежно предшествует стадия ионизации.
4. Электролитическая диссоциация .
Ионизация сольвата является необходимым, но еще не обязательным условием образования электролитного раствора. Обязательным же условием диссоциации ионизированного комплекса на ионы является достаточно высокая диэлектрическая проницаемость раствора. Лишь в этом случае ионизованный комплекс распадается на ионы:

Несмотря на то, что полная ионизация протекает достаточно часто, полная электролитическая диссоциация (сильные электролиты) наблюдается значительно реже. Абсолютно подавляющее большинство изученных электролитных неводных растворов образовано слабыми электролитами, реже — электролитами средней силы.
Основные методы определения константы электролитической диссоциации обычно сводятся к кондукто- либо потенциометрическим методикам и приложимы лишь к весьма разбавленным растворам. Другие методы определения констант диссоциации (например, спектрофотометрические) имеют ограниченное применение.
Современный арсенал методов исследования растворов позволяет почти всегда уверенно определять число стадий для каждого конкретного объекта.