Дипломная работа: Математическое моделирование пластической деформации кристаллов
“К ЗАЩИТЕ”
Заведующий кафедры материалов реакторостроения
Проф*******
ВЫПУСКНАЯ РАБОТА
МАГИСТРА ПРИКЛАДНОЙ ФИЗИКИ
МАТЕМАТИЧЕСКОЕ МОДЕЛИРОВАНИЕ ПЛАСТИЧЕСКОЙ ДЕФОРМАЦИИ И РАЗРУШЕНИЯ ГПУ КРИСТАЛЛОВ
| Руководитель |
************ |
| Студент | ************** |
Харьков
2008
Содержание
| Введение | |
| 1 | Применения МД для исследования пластической деформации кристаллов |
| 1.1 | Ограничения |
| 1.2 | Потенциал |
| 1.3 | Алгоритм интегрирования по времени |
| 1.4 | Процедура минимизации |
| 1.5 | Вычисление сил |
| 1.6 | Периодичность |
| 1.7 | Начальное состояние |
| 1.8 | Начальное состояние для кристалла с дефектами |
| 1.9 | Нагрузка |
| 1.10 | Уравнение для ширины ячейки моделирования |
| 1.11 | Контроль системы |
| 1.12 | Вычисление физических величин |
| 1.13 | Визуализация |
| 2 | Моделирования пластической деформации ГПУ кристаллов |
| Заключение | |
| Список использованных источников |
Анотація
В даній роботі проведено аналіз особливостей застосування методу молекулярної динаміки для моделювання пластичності кристалів. Запропоновано новий підхід до моделювання розтягуваннякристалів. Запропоновано динамічне рівняння для поперечного розміру коміркимоделювання. Створена програма для дослідження процесу пластичної деформації та руйнування кристалів. Проведено моделювання розвитку пластичної деформації ГЩУ кристалів при одноосному розтягуванні. Показана принципова можливість імітації за допомогою цього методу кривих розтягуваннядосконалихкристалів, зміни температури зразка, появи дислокацій, полос ковзання, поодиноких вакансій та їх скупчень, а також процесу руйнування кристалів
Аннотация
В данной работе проведен анализ особенностей применения метода молекулярной динамики для моделирования пластичности кристаллов. Предложен новый подход к моделированию растяжения кристаллов. Предложено динамическое уравнение для поперечного размера ячейки моделирования. Создана программа для исследования процесса пластической деформации и разрушения кристаллов. Проведено моделирование развития пластической деформации ГПУ кристаллов при одноосном растяжении. Показана принципиальная возможность имитации с помощью этого метода кривых растяжения совершенных кристаллов, изменения температуры образца, появления дислокаций, полос скольжения, одиночных вакансий и их скоплений, а также процесса разрушения кристаллов.
Abstract
In the given article the analysis of features of application of a molecular dynamics for simulation of a plasticity of crystals is conducted. The new approach to simulation of strain of crystals is offered. The dynamic equation for transverse dimensions of simulation cell is offered. The code for investigation of the process of a plastic deformation and destruction of crystals is created. The simulation of development of a plastic deformation hcp crystals is carried out at monoaxial expansion. The principal possibility of imitation with the help of this method of stress-strain curves for the perfect crystals, temperature variation of a sample, appearance of dislocations, stripes of slide, single vacancies and their clusters, and also process of crystal destruction.
Введение
Бурный рост ядерной энергетики и широкое развитие работ в области термоядерного синтеза послужили мощным стимулом интенсификации исследований в области облучаемых материалов. Создана новая ветвь материаловедения – атомное, радиационное материаловедение.
Свойства материалов всегда были ключевым звеном, определяющим успех многих инженерных разработок в различных областях техники. Особенно их роль возросла в последнее время при создании сложных конструкций, работающих в экстремальных условиях. Ядерные реакторы, устройства термоядерного синтеза – яркий пример таких конструкций. Сотни различных по составу, структуре и способам изготовления материалов обеспечивают их работоспособность. Но, попадая в условия высоких потоков облучения (до 1020 нейтр/м2 ∙с) и больших флюенсов (до1027 -1028 нейтр/м2 ), они претерпевают значительные структурные перестройки (радиационное повреждение). Следствием этих перестроек является резкое изменение всех физических свойств материалов. Причем эти изменения носят не совсем обычный характер. Ранее ничего подобного не встречалось в обширной практике работ с различными материалами. Так, были обнаружены абсолютно новые явления, происходящие с облученными материалами и сплавами: радиационное охрупчивание, радиационное распухание, радиационное упрочнение, ускоренная диффузия, радиационно-индуцированные фазово-структурные превращения и др.
И этот перечень, судя по всему, будет продолжен, так как исследования в области термоядерного синтеза уже выдвигают новые требования и к конструкционным материалам, которые должны будут работать в еще более жестких условиях (например, выдерживать облучение нейтронами с энергией до 14 МэВ).
Одним из основных факторов, определяющих свойства материалов, как известно, является структура. В условиях облучения она претерпевает существенные перестройки на атомарном уровне. Чтобы выявить принципиальные закономерности поведения материалов в тех или иных условиях эксплуатации, создать материалы с заданными свойствами, необходимо установить связь между изменяющейся атомарной структурой материалов и всей совокупностью их макроскопических свойств. Для решения этой важной задачи радиационного материаловедения привлекаются самые современные физические методы анализа структуры материалов, начиная с уже ставшего традиционным рентгеноструктурного анализа и кончая автоионный микроскопией, оже-спектроскопией и др.
Но в значительном числе случаев все же не удается произвести необходимые структурные изменения. Такая ситуация возможна при известных, но быстро меняющихся внешних условиях (например, ударное воздействие на материал при пролете высокоэнергетических нейтронов). Если же точно не известны изменения в структуре материала на всех этапах какого-либо технологического процесса, то трудно говорить о каком-либо надежном прогнозе его поведения.
В ряде случаев современная аппаратура из-за своего недостаточного разрешения не позволяет наблюдать атомные перестройки в материалах, например отдельные атомные скачки при диффузии, растворении и рост предвыделений второй фазы в сплавах и т.д.
Чтобы преодолеть перечисленные трудности и воссоздать быстроразвивающиеся процессы в материалах, перестройки на атомарном уровне или процессы, когда доступ к материалам ограничен или опасен, все чаще в атомном материаловедении привлекается компьютерный эксперимент. Обзор методов компьютерного моделирования используемых в радиационном материаловедении на начало 90-х годов можно найти в [1].
Метод молекулярной динамики (МД) - один из наиболее мощных методов, используемых для компьютерного моделирования в радиационном материаловедении. Он позволяет проводить детальные исследования структуры материалов исходя из первых принципов. В последние годы метод молекулярной динамики переживает второе рождение. Связано это, во-первых, с быстрым увеличением мощности компьютеров (быстродействия, объема памяти), что позволяет проводить беспрецедентные по масштабам компьютерные эксперименты. Так, сегодня уже проведены расчеты пластической деформации нанокристаллов меди, состоящих из 100 миллионов атомов [2]. С другой стороны, были разработаны более совершенные потенциалы межатомного взаимодействия, включающие в себя также многочастичные эффекты.
Данная работа является продолжением защищенной в 2003 г. бакалаврской работы, в которой были проанализировано современное состояние методов математической обработки кривых растяжения реакторных материалов, а также было проведено, на примере реакторных сталей, определение основных параметров, характеризующих пластическую деформацию.
Целью работы является изучение метода молекулярной динамики и особенностей его применения к исследованию пластичности реакторных материалов, разработка алгоритма и создание программы для изучения пластичности двумерных ГПУ-кристаллов.
1. Применения МД для исследования пластической деформации кристаллов
Для изучения пластических свойств материалов методом МД, атомы из которых состоит материал, помещают в ячейку моделирования, обычно прямоугольной формы, которую подвергают деформации. Координаты и скорости атомов находят, численно решая уравнения движения Ньютона для заданных потенциалов взаимодействия между атомами. Затем, зная координаты и скорости атомов, с помощью соответствующиго усреднения, вычисляют макроскопические величины, такие как температура, давление, тензор напряжения и т.д. Достоинство метода состоит в возможности получить физические величины из первых принципов, т.е. используя только уравнения движения и потенциал межатомного взаимодействия.
1.1. Ограничения
Укажем на ограничения, свойственные этому методу. Во-первых, можно сразу же спросить: почему мы используем законы Ньютона, чтобы двигать атомы, хотя известно, что системы на атомном уровне подчиняются скорее квантовым законам, чем классическим?
Простейшая проверка применимости классического приближения базируется на тепловой длине волны де-Бройля, определяемой как
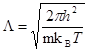 , , | (1) |
где ![]() - атомная масса и
- атомная масса и ![]() - температура. Классическое приближение хорошо работает когда
- температура. Классическое приближение хорошо работает когда ![]() , где
, где ![]() - среднее расстояние между атомами. Если рассматривать, например, жидкость в тройной точке, то
- среднее расстояние между атомами. Если рассматривать, например, жидкость в тройной точке, то ![]() порядка 0.1 для легких элементов, таких как Li и Ar, уменьшаясь для более тяжелых элементов. Классическое приближение плохо работает для легких систем, таких как H2 , He, Ne.
порядка 0.1 для легких элементов, таких как Li и Ar, уменьшаясь для более тяжелых элементов. Классическое приближение плохо работает для легких систем, таких как H2 , He, Ne.
Кроме того, квантовые эффекты становятся важными для любых систем, когда температура ![]() достаточно низка. Падение удельной теплоемкости кристаллов ниже температуры Дебая и аномальное поведение коэффициента теплового расширения есть хорошо известные примеры измеримых квантовых эффектов в твердых телах. Поэтому результаты, полученные с помощью МД должны интерпретироваться с известной осторожностью в этих областях.
достаточно низка. Падение удельной теплоемкости кристаллов ниже температуры Дебая и аномальное поведение коэффициента теплового расширения есть хорошо известные примеры измеримых квантовых эффектов в твердых телах. Поэтому результаты, полученные с помощью МД должны интерпретироваться с известной осторожностью в этих областях.
Второе ограничение связано с ограниченностью используемых компьютерных ресурсов, что приводит к ограничению количества рассматриваемых атомов и, как следствие, к снижению точности вычисляемых физических величин. Частично эту проблему можно обойти, используя подходящие граничные условия (см. ниже). Постоянный рост мощности компьютеров также способствует смягчению этой проблемы. Так, в настоящее время, в печати имеются сообщения о расчетах с 100 млн. атомов.
Необходимо отметить, что современные мощные суперкомпьютеры являются параллельными. Поэтому для расчетов на них необходимо обеспечить эффективное распараллеливание вычислений. Об одной новой возможности распараллеливания в рассматриваемой здесь задаче будет сказано ниже.
Ограниченное быстродействие компьютеров накладывает ограничения на скорость деформации, используемую в вычислениях. Это связано с тем, что при решении уравнений движения шаг по времени должен составлять порядка 0,01 шага от периода колебаний атомов (по порядку величины равного![]() сек) для обеспечения необходимой точности вычислений. Отсюда следует, что для обеспечения деформации ~100% за приемлемое время вычислений типичная скорость деформации должна составлять порядка
сек) для обеспечения необходимой точности вычислений. Отсюда следует, что для обеспечения деформации ~100% за приемлемое время вычислений типичная скорость деформации должна составлять порядка ![]() сек
сек![]() в то время как максимально достигаемая в эксперименте скорость деформации составляет 105 сек-1 . Кроме очевидной возможности достижения более низкой скорости деформации, увеличивая время вычислений, есть еще одна возможность – использовать процедуру минимизации.
в то время как максимально достигаемая в эксперименте скорость деформации составляет 105 сек-1 . Кроме очевидной возможности достижения более низкой скорости деформации, увеличивая время вычислений, есть еще одна возможность – использовать процедуру минимизации.
При нулевой температуре система находится в локальном минимуме внутренней энергии, а из-за отсутствия тепловых колебаний она не может покинуть этот минимум.
Процедура минимизации позволяет деформируемой системе находиться вблизи локального минимума внутренней энергии. При таком моделировании время не определено, так как мы не решаем уравнений движения. Следовательно, скорость деформации не оказывает никакого влияния, если она достаточно низка чтобы исключить нагрев системы, и обеспечить сходимость процедуры минимизации. Таким образом, моделирование, основанное на процедуре минимизации, представляет собой модель идеализированного эксперимента при нулевой температуре в пределе низкой скорости деформации, когда тепло выделяемое при деформировании, удаляется.
1.2. Потенциал
Для моделирования материала необходимо задать потенциал взаимодействия составляющих его атомов. Наиболее простым является парный потенциал Леннарда-Джонса
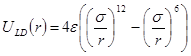 , , | (2) |
Здесь, ![]() - расстояние между атомами,
- расстояние между атомами, ![]() - глубина потенциальной ямы и
- глубина потенциальной ямы и ![]() связано с положением минимума потенциала
связано с положением минимума потенциала ![]() . Потенциал Леннарда-Джонса качественно правильно описывает взаимодействие между атомами – сильное отталкивание на малых расстояниях, обусловленное первым слагаемым в скобках, и притяжение на больших расстояниях, за которое отвечает второе слагаемое в скобках. Он хорошо описывает ван-дер-ваальсовское взаимодействие между атомами кристаллов благородных газов, но, вследствие своей простоты, часто используется для качественного описания взаимодействия других атомов. С потенциалом Леннарда-Джонса проведено большое количество вычислений. Он является стандартным в вычислениях методом МД.
. Потенциал Леннарда-Джонса качественно правильно описывает взаимодействие между атомами – сильное отталкивание на малых расстояниях, обусловленное первым слагаемым в скобках, и притяжение на больших расстояниях, за которое отвечает второе слагаемое в скобках. Он хорошо описывает ван-дер-ваальсовское взаимодействие между атомами кристаллов благородных газов, но, вследствие своей простоты, часто используется для качественного описания взаимодействия других атомов. С потенциалом Леннарда-Джонса проведено большое количество вычислений. Он является стандартным в вычислениях методом МД.
--> ЧИТАТЬ ПОЛНОСТЬЮ <--