Реферат: Синтез похідних 3 аміно 4 оксо 3 4 дигідрохіназоліну на основі антранілоїлгідразиду та дикарбонових

Взаємодія N27 та С12 приводить до утворення структури (2.8), в той же час реакція між N16 та С25 веде до формування семичленного циклу – сполука (2.9). Виходячи з величини зарядів друге перетворення є більш ймовірним, в той час як перше більш можливе з точки зору утворення меншого циклу – шестичленного. Наявність конкуренції між цими процесами, на нашу думку, і є причиною утворення двох продуктів даної реакції.
З аналізу ЯМР 1 Н-спектрів одним із продуктів реакції є N-2,3-дибензил-4,10-діоксо-4,10-дигідро-3Н -[1,2, 4]триазіно[6,1-b ]хіназолін-2-карбоксамід (2.8), на що вказує наявність триплетного сигналу NH-протону при 9,35 м.ч. та дублетного сигналу СН2 -групи бензиламідного фрагменту при 4,32 м.ч. Про утворення структури (2.9) в ЯМР-спектрі свідчать уширений синглетний сигнал NH‑протону (12,67 м.ч.), а також присутні сигнали АBCD-системи хіназолонового фрагменту в області 7,60...8,23 м.ч. та протонів бензенових ядер у вигляді складного мультиплету при 7,10...7,45 м.ч.
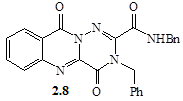 |  |
Циклізацією діестеру (2.2) здійснений синтез 2-карбетокси-3-N-етоксаліл-аміно-4-оксо-3,4-дигідрохіназоліна (2.10).
2. Синтез пох і дних 3-аміно-4-оксо-3,4-дигідрохіназоліну на основі антранілогідразиду та дикарбонових кислот
Послідовним ацилюванням антранілогідразиду (2.1) янтарним ангідридом та етоксалілхлоридом в льодяній оцтовій кислоті в присутності триетиламіну одержували естерокислоту (2.12). При нагріванні естерокислота (2.12) в оцтовій кислоті циклізувалася в похідне хіназолінону (2.13), обробка якого оцтовим ангідридом приводила до цільового іміду (2.14). Останній отримували також при дії оцтового ангідриду на сполуку (2.12); реакцію проводили без виділення проміжного продукту (2.13).
Імід (2.14), окрім сукцинімідного циклу містить в своєму складі естерну групу, обидві вони, як відомо, легко вступають в реакції з N– і О–нуклеофілами. В реакції іміду (2.14) з алкіламінами були виділені відповідні аміди (2.15 а-в), що свідчить про більшу реакційну здатність імідного циклу порівняно з естерною групою.
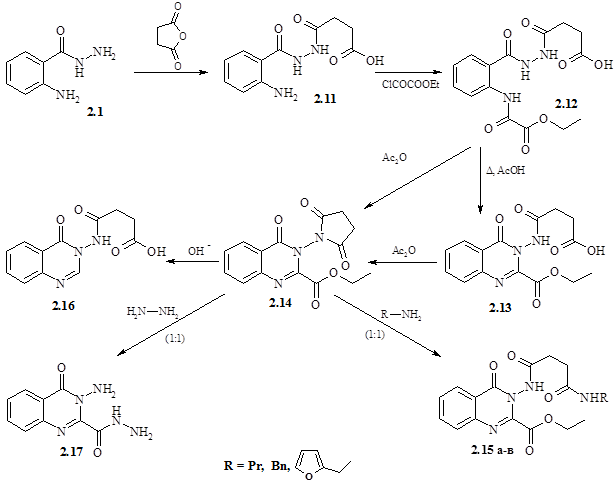
Проведені розрахунки електронної густини методом АМ1 на атомах вуглецю карбонільних груп показали, що вони відповідно становлять +0.428 на атомі вуглецю естерної групи і +0.394 і +0.378 на карбонільних атомах вуглецю імідного ядра. Це свідчить на користь більш високої реакційної здатності естерної групи по відношенню до імідного циклу. Різниця в зарядах на карбонільних атомах вуглецю імідного ядра обумовлена, скоріш за все, порушенням спряження в ньому. Аналогічну закономірність одержали і при розрахунках іншими методами (MINDO/3, MNDO, PM3). Те, що реакція амідування протікає не за естерною групою, а по імідному кільцю можна пояснити просторовими факторами.
В реакції іміду (2.14) з гідразингідратом був виділений один з продуктів реак-ції – гідразид (2.17) з виходом 30%. Ймовірно, на початку реакції проходить, як і у випадку реакції амідування, розкриття імідного циклу а далі відщеплюється залишок янтарної кислоти. В силу більшої нуклеофільності гідразину паралельно перебігає й реакція гідразинолізу за естерною групою. При обробці іміду (2.14) надлишком лугу з реакційного середовища була виділена кислота (2.16), продукт декарбоксилювання по положенню 2.
Наявність в молекулі антранілогідразиду фталевої кислоти поряд із залишками інших дикарбонових кислот є вирішальним фактором напрямку реакції; багато чого залежить і від того, яку саме групу проацильовано фталевим ангідридом. Якщо залишок фталевої кислоти знаходиться в гідразидному фрагменті молекули, то в реакції спостерігається замикання в першу чергу фталімідного цикла – сполуки (2.20, 2.21). Так, при ацилюванні кислоти (2.19) етоксалілхлоридом на холоді одержували імід (2.20). Якщо реакцію проводити при нагріванні, як у випадку ацилювання кислоти (2.19) янтарним ангідридом, то утворюється імідокислота (2.21). Фталева кислота досить легко (порівняно з іншими дикарбоновими кислотами) утворює імідний цикл, а крім того гідразидна група також сприяє утворенню цього циклу в силу її підвищених нуклеофільних властивостей (a–ефект).
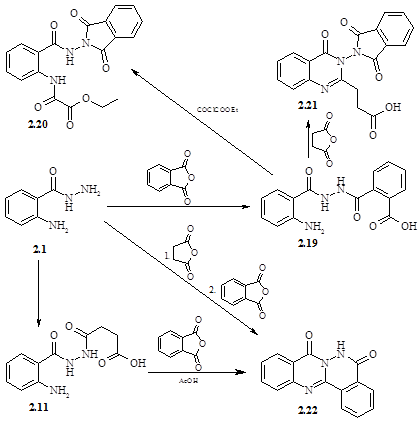
Напрямок реакції змінюється якщо фталевим ангідридом проацильована аміногрупа антранілогідразиду; так, ацилювання кислоти (2.11) фталевим ангідридом і наступним кип‘ятінням в оцтовій кислоті веде до утворення хіназолінону (2.22). Останній був також отриманий при послідовному ацилюванні гідразида (2.1) янтарним і фталевим ангідридами в оцтовій кислоті. Утворення хіназолінону (2.22) є результатом першочергового замикання хіназолінонового циклу, а наступне замикання фталазинового циклу супроводжується одночасним витисненням залишку янтарної кислоти.
3 . Синтез 3-сукцинімідо-4-оксо-3,4-дигідрохіназоліну і похідних N‑(4‑оксохіназолін-3-іл)карбамоїлпропанової кислоти на його основі
Взаємодією антранілогідразида (2.1) з діетилоксалатом в умовах мікрохвильового опромінення замість очікуваного 3-аміно-2-етоксикарбоніл-4-оксо-3,4-дигідрохиназоліну був одержаний 3-аміно-4-оксо-3,4-дигідрохіназолін (3.2). В спектрі ЯМР 1 Н сполуки (3.2) фіксувалися вузький сінглет протонів групи NH2 (d 5.88 м.д.) і сигнал Н2 хіназолінонового ядра (d 8.37 м.д.), а сигнали протонів алкільного радикала були відсутні. Запропоновано можливі шляхи утворення сполуки (3.2).
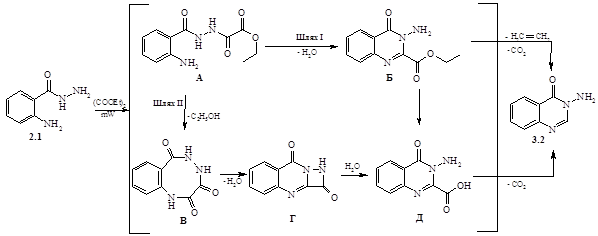
Найімовірніше, спочатку відбувається ацилування гідразида (2.1) діетилоксалатом з утворенням сполуки (А), яка за одним із шляхів (шлях І) в умовах реакції переацилується, утворюючи естер (Б). Декарбоксилування останнього приводить до кінцевого продукту реакції (3.2). За іншим ймовірним шляхом (шлях ІІ) сполука А зазнає внутрішньо-молекулярного ацилювання з утворенням інтермедіату В, який через проміжні продукти Г, Д перетворюється в хіназолінон (3.2). З метою експериментальної перевірки можливості протікання реакції по першому шляху нами була синтезована сполука Б, яку ми спробували декарбоксилювати в умовах реакції. Виявилось, що навіть при більш жорстких умовах (збільшення потужності опромінення і часу перебігу до 10 хв.) була виділена вихідна речовина, тому більш вірогідним із запропонованих можна вважати шлях II.
Для подальших досліджень хіназолон (3.2) одержували нагріванням антранілогідразида (2.1) в середовищі мурашиної кислоти.

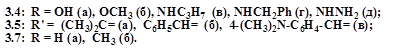
При недовготривалому кип’ятінні хіназолінона (3.2) з янтарним ангідридом в крижаній оцтовій кислоті, був виділений 3-сукцинімідо-4-оксо-3,4-дигідро-хіназолін (3.3). Реакція перебігає з утворенням проміжної сполуки – кислоти (3.4a), яка в результаті циклодегідратації перетворюється в імід (3.3), при цьому відмічаємо легкість, з якою відбувається замикання імідного циклу. Навіть слабке нагрівання хіназолінону (3.2) з янтарним ангідридом дає суміш кислоти (3.4a) і іміда (3.3) з перевагою в ній останнього. При проведенні реакції ацилювания без нагрівання (перемішування, 48 год.) з реакційної суміші знову була виділена суміш вихідної речовини (3.2) і кислоти (3.4a). Тому одержувати N-4-оксо-3,4-дигідрохіназолін-3-іл)карбомоїл-пропанову кислоту (3.4a) було запропоновано лужним гідролізом іміду (3.3).
Імід (3.3) легко вступає в SN -реакції: при кип’ятінні його в метанолі з метилатом натрію був виділений естер (3.4б), а при нагріванні з аліфатичними амінами в етанолі було одержано N-заміщені аміди N′-(4-оксо-3,4-дигідрохіназолін-3-іл)карбамоїлпропанової кислоти (3.4в,г). Взаємодією іміда (3.3) з гідразингідратом в середовищі діоксану синтезовано гідразид (3.4д). При кип’ятінні гідразида (3.4д) з ацетоном або ароматичними альдегідами в середовищі ДМФА були виділені відповідні гідразони (3.5а-в).
Для синтезу біспохідних 4-оксохіназоліна (3.7а,б) використовували сполуку (3.6), яку одержували нагріванням іміда (3.3) і гідразида (2.1) в середовищі діоксану або сплавленням вихідних речовин. Кращі результати спостерігалися при проведенні реакції в розчиннику. Циклізацією фрагмента антранілогідразида в молекулі сполуки (3.6) отримували відповідні біспохідні. При нагріванні сполуки (3.6) в середовищі мурашиної кислоти утворюється N,N′-ди(4-оксо-3,4-дигідрохіназолін-3-іл)сукцинамід (3.7а). Крім того, сукцинамід (3.7a) був також одержаний сплавленням іміда (3.3) з хіназоліноном (3.2). В цьому випадку реакція перебігала з більш низьким виходом, а підвищення температури або часу нагрівання призводило до руйнування речовин. Спроба провести реакцію між сполуками (3.2) і (3.3) в розчиннику успіху не мала: вихідні речовини були виділені при кип’ятінні реагентів (3.2) і (3.3) в етанолі, діоксані, оцтовій кислоті і в ДМФА. При нагріванні сполуки (3.6) з еквімолярною кількістю оцтового ангідриду в середовищі крижаної оцтової кислоти отримано біспохідне (3.7б).
4. Реакції етилового естеру оксамінової кислоти і діалкілоксалатів з похідними антранілової кислоти
В літературі описаний синтез етилового естеру 3-аміно-4-оксо-3,4-дигідрохіназолін-2-карбонової кислоти (3.9а) нагріванням антранілогідразида (2.1) з діетилоксалатом при 180ºС потягом 6 годин і подальшою відгонкою діетилоксалату у вакуумі. Ми спробували одержати амід 3-аміно-4-оксо-3,4-дигідрохіназолін-2-карбонової кислоти взаємодією антранілогідразиду (2.1) з етиловим естером оксамінової кислоти (оксаметаном), в якості розчинника використовували оцтову кислоту. Результат реакції виявився несподіваним – із реакційної суміші був виділений етиловий естер (3.9а), тобто оксаметан реагував не естерною, а амідною групою. Використання подвійної кількості оксаметана в цій реакції не змінило її результат і не впливало на вихід продукту реакції. Вперше було виявлено реакцію естерів оксамінової кислоти з N-нуклеофілами, яка протікає за амідною групою, без участі естерної.
При нагріванні кислоти (3.8б) і антраніламіда (3.8г) з оксаметаном були одержані етилові естери 2-карбоксіоксанілової (3.10) та 4-оксо-3,4-дигідрохіназолін-2-карбонової кислот (3.9в) відповідно, тобто оксаметан і в цих випадках вступав в реакцію амідним фрагментом молекули.
У випадку з метилантранілатом (3.8в) результат реакції змінився і було виділено амід2-метоксикарбонілоксанілової кислоти (3.11), тобто більш реакційноздатною виявилась естерна група оксаметану. Можна припустити, що вирішальним фактором, який визначає напрямок реакції утворення аміда (3.11), є відсутність в метоксикарбонільній групі метилантранілата (3.8в) атомів водню, здатних утворювати внутрішньомолекулярні водневі зв’язки, як у випадку антранілової кислоти (3.8б) і інших її похідних (3.8а,г). Очевидно, водневі зв’язки впливають на формування структури перехідного комплексу та природу групи, що відщеплюється.
Дослідження реакції ацилювання похідних антранілової кислоти діалкілоксалатами дозволило розробити менш трудомісткий метод синтезу естерів (3.9а,б).
Етиловий і метиловий естери3-аміно-4-оксо-3,4-дигідрохіназолін-2-карбонової кислоти (3.9а) та (3.9б) відповідно отримували кип’ятінням антранілогідразиду (2.1) і відповідного діалкілоксалату (Alk = Et або Ме) протягом 30 хв. Разом з тим, виділити естери з іншими алкільними залишками (в реакції були використані (COОAlk)2 , де Alk = н- Pr, н- Bu, н- C6 H13 , н- С5 Н11 ) в цих умовах нам не вдалося, навіть при збільшенні часу перебігу реакції. Вірогідно, це пов’язано з більш низькою реакційною здатністю таких естерів. Реакція антранілогідразиду (2.1) з етоксалілхлоридом в оцтовій кислоті в присутності триетиламіну давала 3‑N-ацетиламіно-2-карбетокси-4-оксо-3,4-дигідрохіназолін (3.13).

3.8, X = NHNH2 (а – 2.1), OH (б), OMe (в), NH2 (г); 3.9, R = NH2 , Alk = Et (a), Me (б), R = H, Alk = Et (в), Me (г);3.12, Alk = Et (a), Me (б); 3.14, R = H (a), NH2 (б).
В крижаній оцтовій кислоті антраніламід (3.8г) з діетилоксалатом в залежності від часу протікання реакції дає естер (3.9в) і етиловий естер 2‑карбамоїлоксанілової кислоти (3.12а) або їх суміш, при використанні диметилоксалату в реакції утворювався метиловий естер 4-оксо-3,4-ди-гідрохіназолін-2-карбонової кислоти (3.9г).